Welch Joshua D, Baran-Gale Jeanette, Perou Charles M, Sethupathy Praveen, Prins Jan F
Curriculum in Bioinformatics and Computational Biology, The University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
Department of Computer Science, The University of North Carolina at Chapel Hill, Chapel Hill, NC, 27599, USA.
BMC Genomics. 2015 Feb 22;16(1):113. doi: 10.1186/s12864-015-1227-8.
Recent studies have shown that some pseudogenes are transcribed and contribute to cancer when dysregulated. In particular, pseudogene transcripts can function as competing endogenous RNAs (ceRNAs). The high similarity of gene and pseudogene nucleotide sequence has hindered experimental investigation of these mechanisms using RNA-seq. Furthermore, previous studies of pseudogenes in breast cancer have not integrated miRNA expression data in order to perform large-scale analysis of ceRNA potential. Thus, knowledge of both pseudogene ceRNA function and the role of pseudogene expression in cancer are restricted to isolated examples.
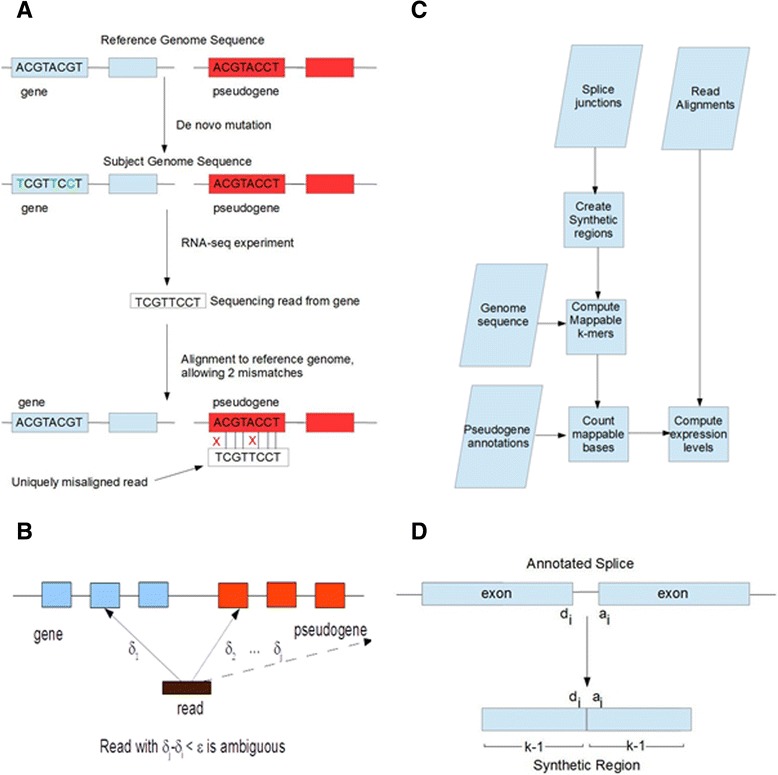
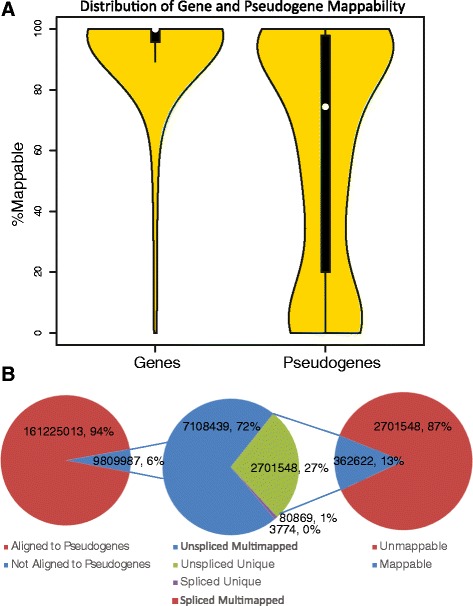
To investigate whether transcribed pseudogenes play a pervasive regulatory role in cancer, we developed a novel bioinformatic method for measuring pseudogene transcription from RNA-seq data. We applied this method to 819 breast cancer samples from The Cancer Genome Atlas (TCGA) project. We then clustered the samples using pseudogene expression levels and integrated sample-paired pseudogene, gene and miRNA expression data with miRNA target prediction to determine whether more pseudogenes have ceRNA potential than expected by chance.
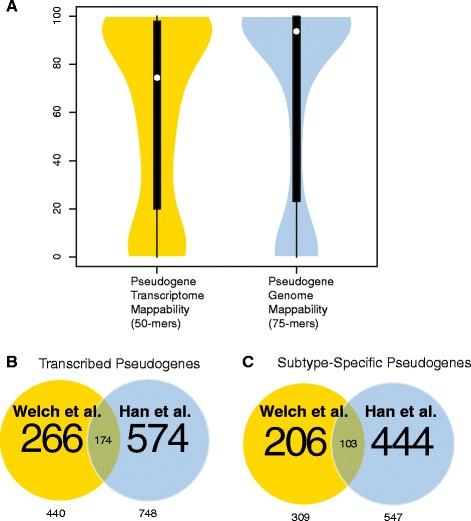
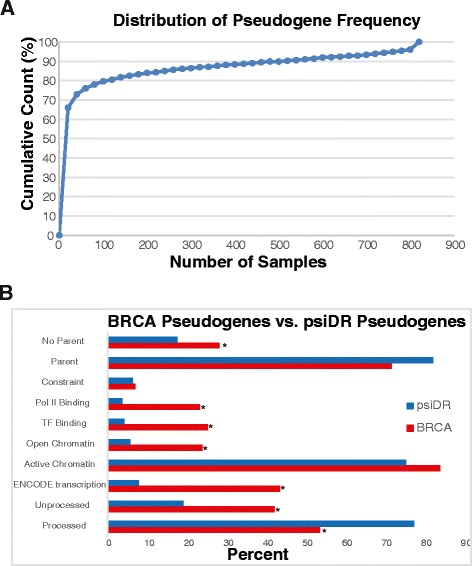
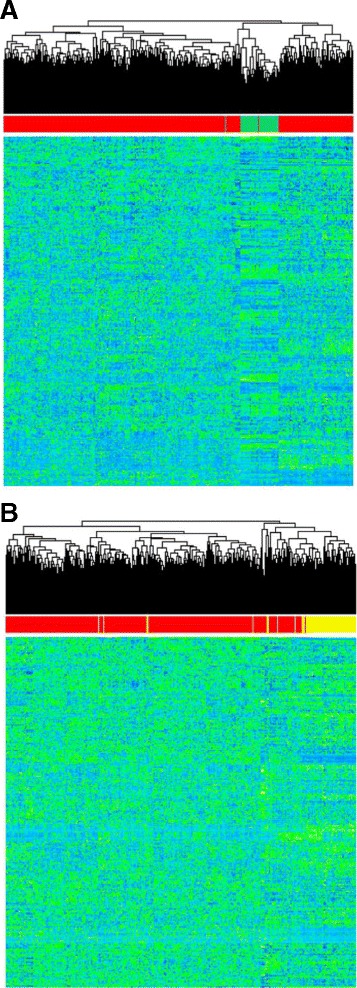
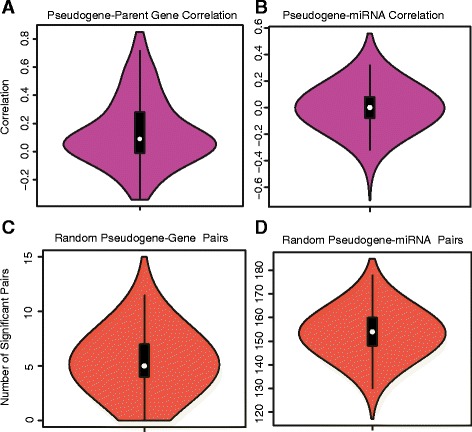
Our analysis identifies with high confidence a set of 440 pseudogenes that are transcribed in breast cancer tissue. Of this set, 309 pseudogenes exhibit significant differential expression among breast cancer subtypes. Hierarchical clustering using only pseudogene expression levels accurately separates tumor samples from normal samples and discriminates the Basal subtype from the Luminal and Her2 subtypes. Correlation analysis shows more positively correlated pseudogene-parent gene pairs and negatively correlated pseudogene-miRNA pairs than expected by chance. Furthermore, 177 transcribed pseudogenes possess binding sites for co-expressed miRNAs that are also predicted to target their parent genes. Taken together, these results increase the catalog of putative pseudogene ceRNAs and suggest that pseudogene transcription in breast cancer may play a larger role than previously appreciated.
近期研究表明,一些假基因会被转录,且在失调时会促进癌症发生。特别是,假基因转录本可作为竞争性内源RNA(ceRNA)发挥作用。基因与假基因核苷酸序列的高度相似性阻碍了利用RNA测序对这些机制进行实验研究。此外,先前关于乳腺癌假基因的研究并未整合miRNA表达数据,无法对ceRNA潜力进行大规模分析。因此,关于假基因ceRNA功能以及假基因表达在癌症中的作用的认识仅限于个别例子。
为了研究转录的假基因在癌症中是否发挥普遍的调控作用,我们开发了一种新的生物信息学方法,用于从RNA测序数据中测量假基因转录。我们将此方法应用于来自癌症基因组图谱(TCGA)项目的819份乳腺癌样本。然后,我们利用假基因表达水平对样本进行聚类,并将样本配对的假基因、基因和miRNA表达数据与miRNA靶标预测整合起来,以确定具有ceRNA潜力的假基因数量是否比随机预期的更多。
我们的分析高度可靠地鉴定出一组在乳腺癌组织中被转录的440个假基因。在这组假基因中,有309个在乳腺癌亚型之间表现出显著的差异表达。仅使用假基因表达水平进行层次聚类就能准确地将肿瘤样本与正常样本区分开,并将基底亚型与管腔型和Her2亚型区分开。相关性分析表明,假基因-亲本基因对呈正相关以及假基因-miRNA对呈负相关的情况比随机预期的更多。此外,177个转录的假基因拥有与共表达miRNA的结合位点,这些miRNA也被预测靶向它们的亲本基因。综上所述,这些结果增加了假定的假基因ceRNA目录,并表明乳腺癌中的假基因转录可能比之前认为的发挥更大的作用。