Department of Biology, Center for Computational and Integrative Biology, Rutgers University , Camden, NJ , USA.
PeerJ. 2015 Mar 17;3:e836. doi: 10.7717/peerj.836. eCollection 2015.
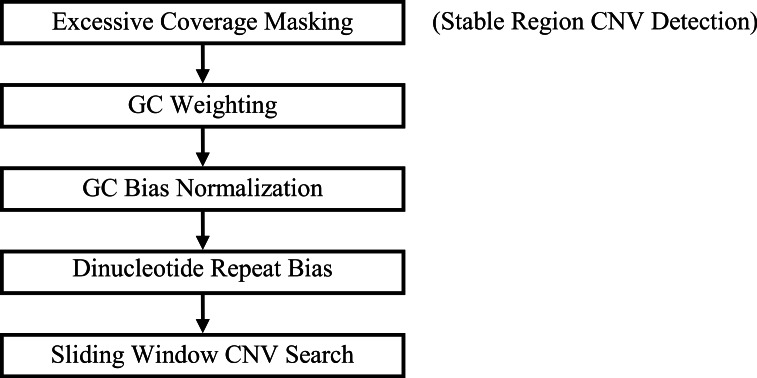
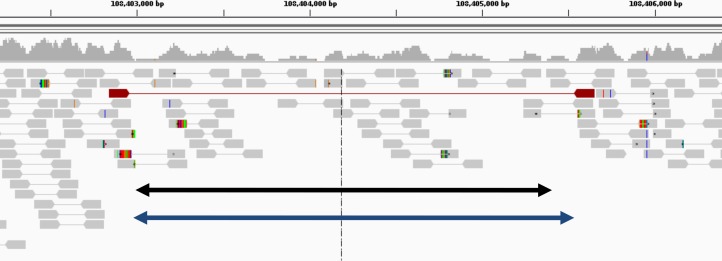
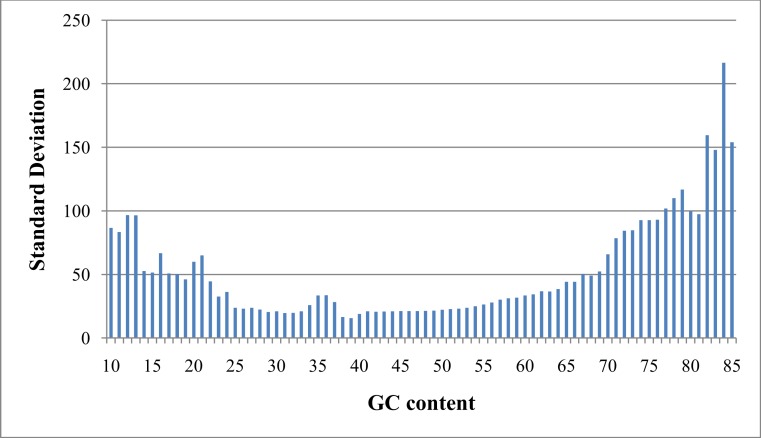
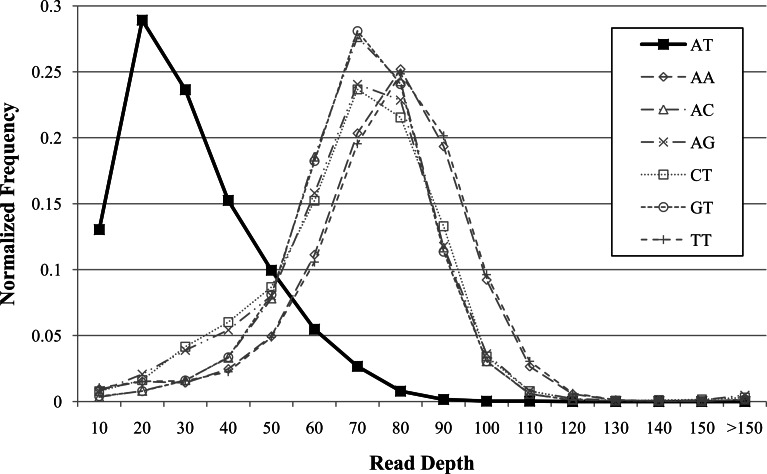
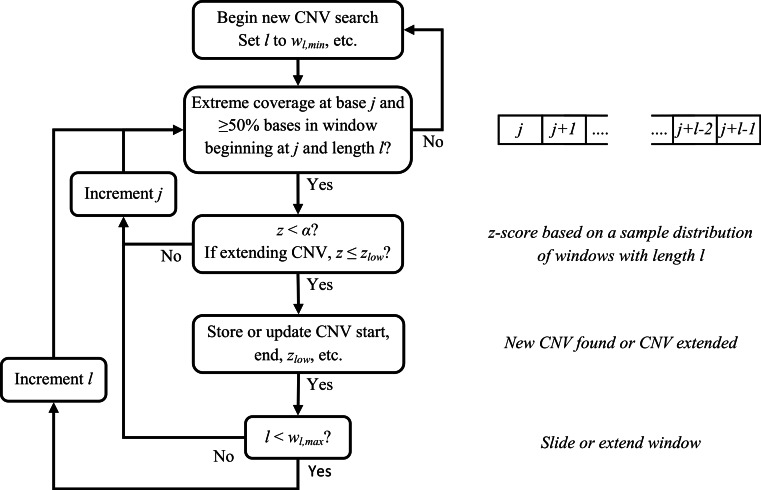
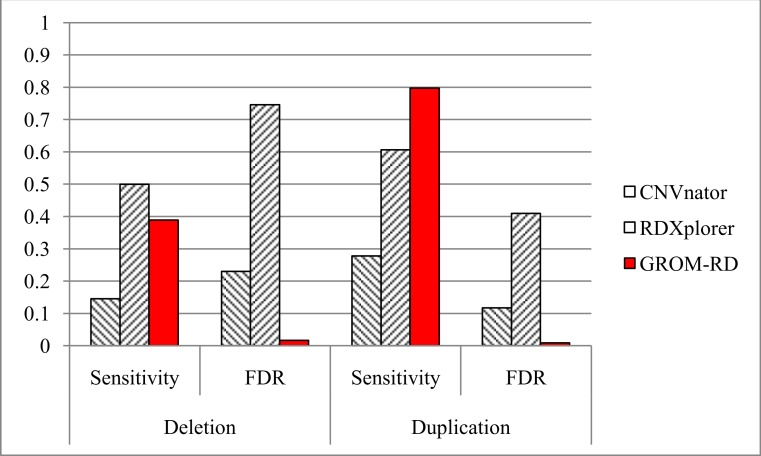
Amplifications or deletions of genome segments, known as copy number variants (CNVs), have been associated with many diseases. Read depth analysis of next-generation sequencing (NGS) is an essential method of detecting CNVs. However, genome read coverage is frequently distorted by various biases of NGS platforms, which reduce predictive capabilities of existing approaches. Additionally, the use of read depth tools has been somewhat hindered by imprecise breakpoint identification. We developed GROM-RD, an algorithm that analyzes multiple biases in read coverage to detect CNVs in NGS data. We found non-uniform variance across distinct GC regions after using existing GC bias correction methods and developed a novel approach to normalize such variance. Although complex and repetitive genome segments complicate CNV detection, GROM-RD adjusts for repeat bias and uses a two-pipeline masking approach to detect CNVs in complex and repetitive segments while improving sensitivity in less complicated regions. To overcome a typical weakness of RD methods, GROM-RD employs a CNV search using size-varying overlapping windows to improve breakpoint resolution. We compared our method to two widely used programs based on read depth methods, CNVnator and RDXplorer, and observed improved CNV detection and breakpoint accuracy for GROM-RD. GROM-RD is available at http://grigoriev.rutgers.edu/software/.
基因组片段的扩增或缺失,即拷贝数变异(CNV),与许多疾病有关。下一代测序(NGS)的读深度分析是检测 CNV 的一种重要方法。然而,NGS 平台的各种偏差经常会扭曲基因组读的覆盖度,从而降低现有方法的预测能力。此外,由于精确的断点识别不精确,读深度工具的使用受到了一定的阻碍。我们开发了 GROM-RD,这是一种分析读覆盖度中的多种偏差以检测 NGS 数据中 CNV 的算法。在使用现有的 GC 偏差校正方法后,我们发现不同 GC 区域的方差不均匀,并开发了一种新的方法来归一化这种方差。尽管复杂和重复的基因组片段使 CNV 检测变得复杂,但 GROM-RD 可以调整重复偏差,并使用双流水线掩蔽方法来检测复杂和重复片段中的 CNV,同时提高在较简单区域的灵敏度。为了克服 RD 方法的一个典型弱点,GROM-RD 使用大小变化的重叠窗口进行 CNV 搜索,以提高断点分辨率。我们将我们的方法与基于读深度方法的两种广泛使用的程序 CNVnator 和 RDXplorer 进行了比较,发现 GROM-RD 可以提高 CNV 检测和断点的准确性。GROM-RD 可在 http://grigoriev.rutgers.edu/software/ 获得。