Krüger Janine, Brachs Sebastian, Trappiel Manuela, Kintscher Ulrich, Meyborg Heike, Wellnhofer Ernst, Thöne-Reineke Christa, Stawowy Philipp, Östman Arne, Birkenfeld Andreas L, Böhmer Frank D, Kappert Kai
Center for Cardiovascular Research/CCR, Institute of Laboratory Medicine, Clinical Chemistry and Pathobiochemistry, Hessische Str. 3-4, 10115 Berlin, Charité - Universitätsmedizin Berlin, Germany.
Center for Cardiovascular Research/CCR, Department of Endocrinology, Diabetes and Nutrition, Hessische Str. 3-4, 10115 Berlin, Charité - Universitätsmedizin Berlin, Germany.
Mol Metab. 2015 Feb 12;4(4):325-36. doi: 10.1016/j.molmet.2015.02.001. eCollection 2015 Apr.
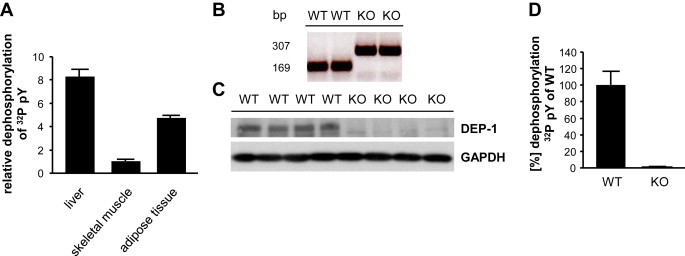
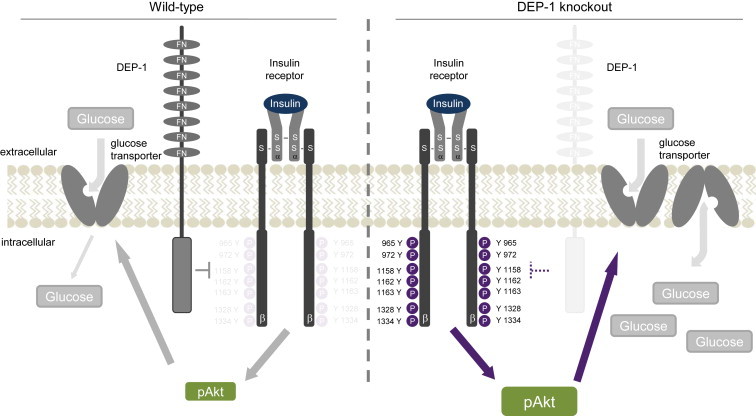
Insulin resistance can be triggered by enhanced dephosphorylation of the insulin receptor or downstream components in the insulin signaling cascade through protein tyrosine phosphatases (PTPs). Downregulating density-enhanced phosphatase-1 (DEP-1) resulted in an improved metabolic status in previous analyses. This phenotype was primarily caused by hepatic DEP-1 reduction.
Here we further elucidated the role of DEP-1 in glucose homeostasis by employing a conventional knockout model to explore the specific contribution of DEP-1 in metabolic tissues. Ptprj (-/-) (DEP-1 deficient) and wild-type C57BL/6 mice were fed a low-fat or high-fat diet. Metabolic phenotyping was combined with analyses of phosphorylation patterns of insulin signaling components. Additionally, experiments with skeletal muscle cells and muscle tissue were performed to assess the role of DEP-1 for glucose uptake.
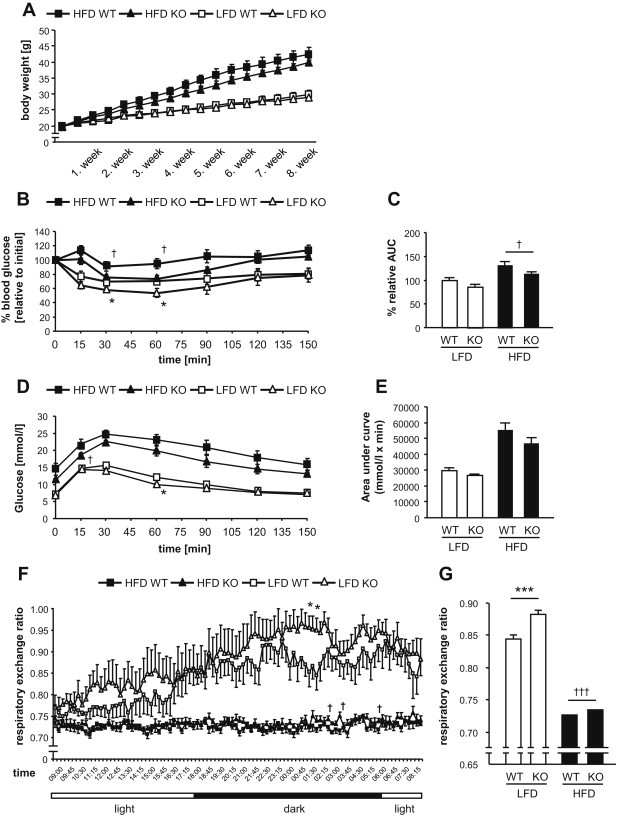
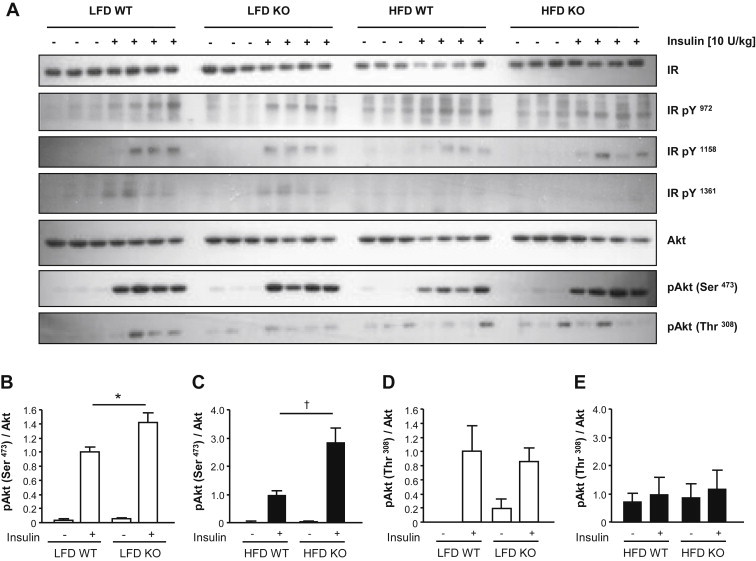
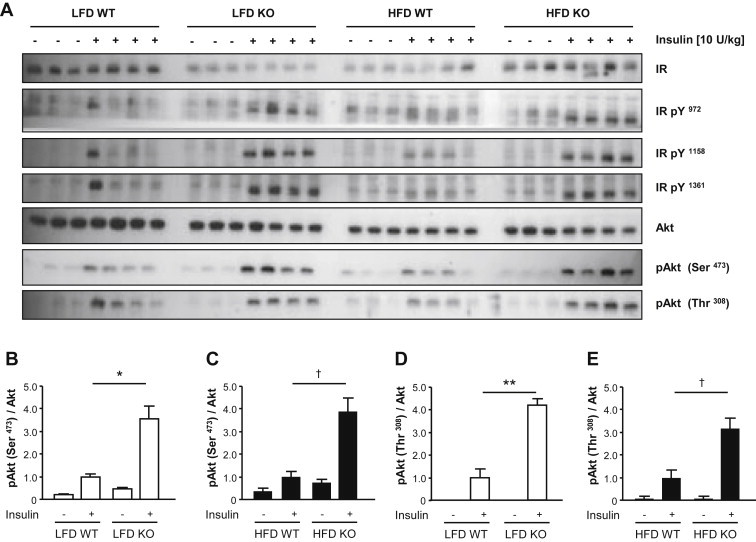
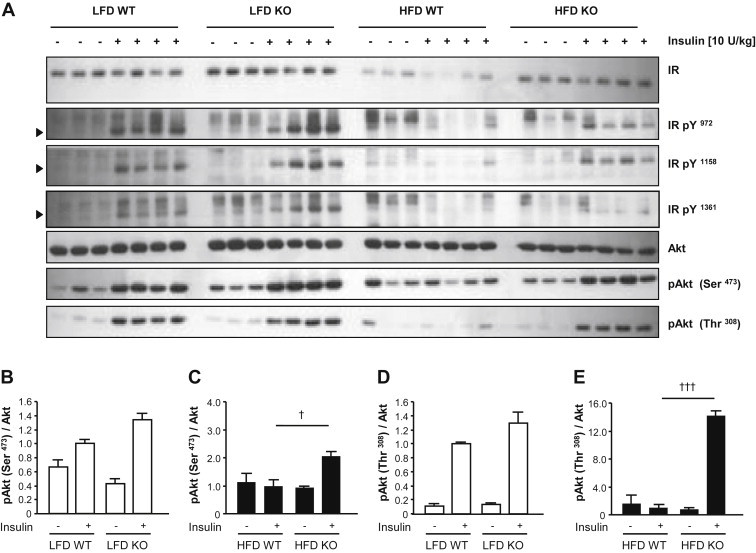
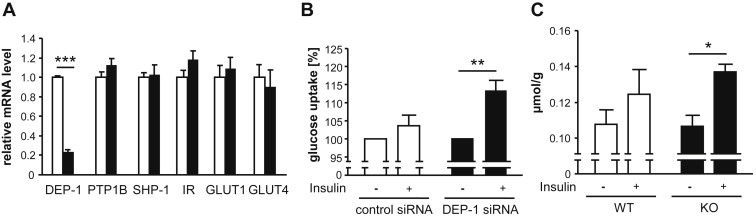
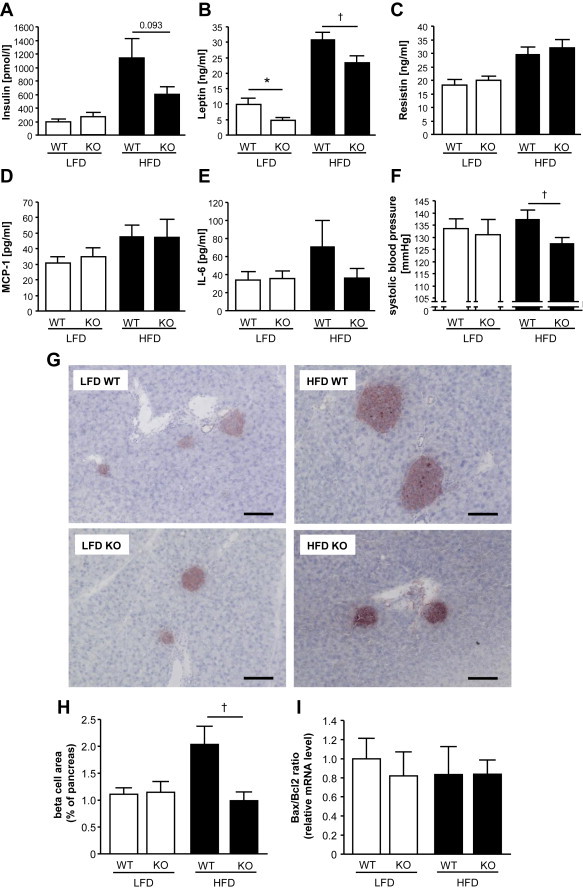
High-fat diet fed-Ptprj (-/-) mice displayed enhanced insulin sensitivity and improved glucose tolerance. Furthermore, leptin levels and blood pressure were reduced in Ptprj (-/-) mice. DEP-1 deficiency resulted in increased phosphorylation of components of the insulin signaling cascade in liver, skeletal muscle and adipose tissue after insulin challenge. The beneficial effect on glucose homeostasis in vivo was corroborated by increased glucose uptake in skeletal muscle cells in which DEP-1 was downregulated, and in skeletal muscle of Ptprj (-/-) mice.
Together, these data establish DEP-1 as novel negative regulator of insulin signaling.
胰岛素抵抗可由胰岛素受体或胰岛素信号级联反应下游成分通过蛋白酪氨酸磷酸酶(PTPs)的去磷酸化增强所引发。在先前的分析中,下调密度增强磷酸酶-1(DEP-1)可改善代谢状态。这种表型主要是由肝脏中DEP-1的减少引起的。
在此,我们通过采用传统的基因敲除模型进一步阐明DEP-1在葡萄糖稳态中的作用,以探究DEP-1在代谢组织中的具体贡献。给Ptprj(-/-)(DEP-1缺陷型)和野生型C57BL/6小鼠喂食低脂或高脂饮食。将代谢表型分析与胰岛素信号成分的磷酸化模式分析相结合。此外,还进行了骨骼肌细胞和肌肉组织的实验,以评估DEP-1对葡萄糖摄取的作用。
喂食高脂饮食的Ptprj(-/-)小鼠表现出增强的胰岛素敏感性和改善的葡萄糖耐量。此外,Ptprj(-/-)小鼠的瘦素水平和血压降低。DEP-1缺陷导致胰岛素刺激后肝脏、骨骼肌和脂肪组织中胰岛素信号级联反应成分的磷酸化增加。在DEP-1被下调的骨骼肌细胞以及Ptprj(-/-)小鼠的骨骼肌中,葡萄糖摄取增加,这证实了DEP-1缺陷对体内葡萄糖稳态的有益作用。
总之,这些数据确定DEP-1是胰岛素信号的新型负调节因子。