Winslow Sofia, Leandersson Karin, Edsjö Anders, Larsson Christer
Breast Cancer Res. 2015 Feb 21;17(1):23. doi: 10.1186/s13058-015-0530-2.
Global gene expression analysis of tumor samples has been a valuable tool to subgroup tumors and has the potential to be of prognostic and predictive value. However, tumors are heterogeneous, and homogenates will consist of several different cell types. This study was designed to obtain more refined expression data representing different compartments of the tumor.
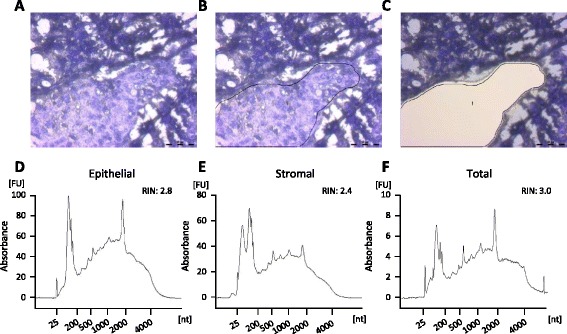
Formalin-fixed paraffin-embedded stroma-rich triple-negative breast cancer tumors were laser-microdissected, and RNA was extracted and processed to enable microarray hybridization. Genes enriched in stroma were identified and used to generate signatures by identifying correlating genes in publicly available data sets. The prognostic implications of the signature were analyzed.
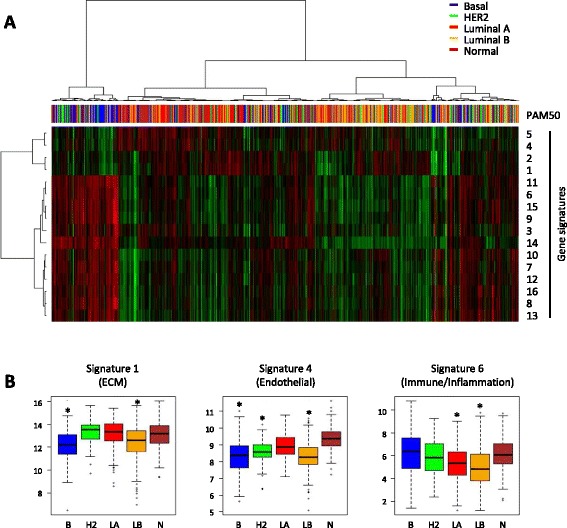
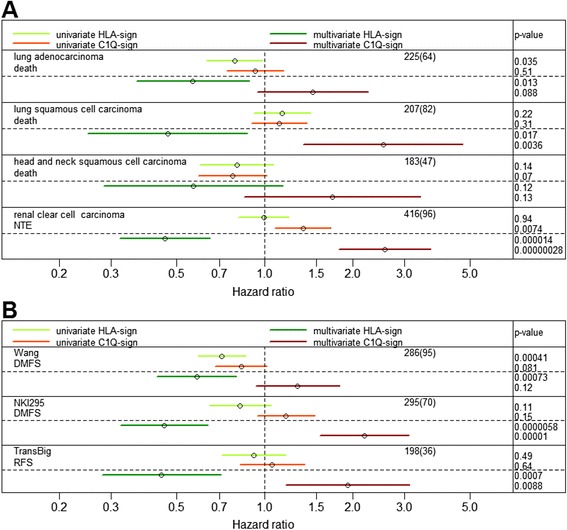
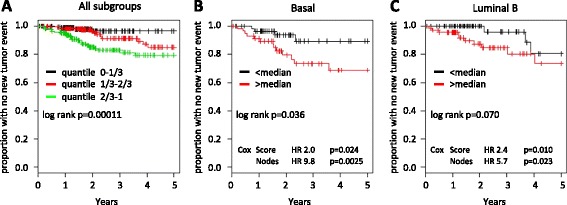
Comparison of the expression pattern from stromal and cancer cell compartments from three tumors revealed a number of genes that were essentially specifically expressed in the respective compartments. The stroma-specific genes indicated contribution from fibroblasts, endothelial cells, and immune/inflammatory cells. The gene set was expanded by identifying correlating mRNAs using breast cancer mRNA expression data from The Cancer Genome Atlas. By iterative analyses, 16 gene signatures of highly correlating genes were characterized. Based on the gene composition, they seem to represent different cell types. In multivariate Cox proportional hazard models, two immune/inflammatory signatures had opposing hazard ratios for breast cancer recurrence also after adjusting for clinicopathological variables and molecular subgroup. The signature associated with poor prognosis consisted mainly of C1Q genes and the one associated with good prognosis contained HLA genes. This association with prognosis was seen for other cancers as well as in other breast cancer data sets.
Our data indicate that the molecular composition of the immune response in a tumor may be a powerful predictor of cancer prognosis.
肿瘤样本的全基因表达分析一直是对肿瘤进行亚组分类的重要工具,并且具有预后和预测价值。然而,肿瘤具有异质性,匀浆将由几种不同的细胞类型组成。本研究旨在获取更精确的表达数据,以代表肿瘤的不同部分。
对福尔马林固定石蜡包埋的富含基质的三阴性乳腺癌肿瘤进行激光显微切割,提取RNA并进行处理以进行微阵列杂交。通过在公开可用的数据集中鉴定相关基因,确定富集于基质中的基因并用于生成特征基因集。分析该特征基因集的预后意义。
对三个肿瘤的基质和癌细胞部分的表达模式进行比较,发现了一些在各自部分基本特异性表达的基因。基质特异性基因表明成纤维细胞、内皮细胞和免疫/炎症细胞的作用。利用来自癌症基因组图谱的乳腺癌mRNA表达数据鉴定相关mRNA,从而扩展了基因集。通过迭代分析,鉴定出16个高度相关基因的特征基因集。根据基因组成,它们似乎代表不同的细胞类型。在多变量Cox比例风险模型中,即使在调整临床病理变量和分子亚组后,两个免疫/炎症特征基因集对乳腺癌复发的风险比也相反。与预后不良相关的特征基因集主要由C1Q基因组成,与预后良好相关的特征基因集包含HLA基因。在其他癌症以及其他乳腺癌数据集中也观察到了这种与预后的关联。
我们的数据表明,肿瘤中免疫反应的分子组成可能是癌症预后的有力预测指标。