Raha Paromita, Thomas Scott, Thurn K Ted, Park Jeenah, Munster Pamela N
Breast Cancer Res. 2015 Feb 25;17(1):26. doi: 10.1186/s13058-015-0533-z.
The emergence of hormone therapy resistance, despite continued expression of the estrogen receptor (ER), is a major challenge to curing breast cancer. Recent clinical studies suggest that epigenetic modulation by histone deacetylase (HDAC) inhibitors reverses hormone therapy resistance. However, little is known about epigenetic modulation of the ER during acquired hormone resistance. Our recent phase II study demonstrated that HDAC inhibitors re-sensitize hormone therapy-resistant tumors to the anti-estrogen tamoxifen. In this study, we sought to understand the mechanism behind the efficacy of this combination.
We generated cell lines resistant to tamoxifen, named TAMRM and TAMRT, by continuous exposure of ER-positive MCF7 and T47D cells, respectively to 4-hydroxy tamoxifen for over 12 months. HDAC inhibition, along with pharmacological and genetic manipulation of key survival pathways, including ER and Bcl-2, were used to characterize these resistant models.
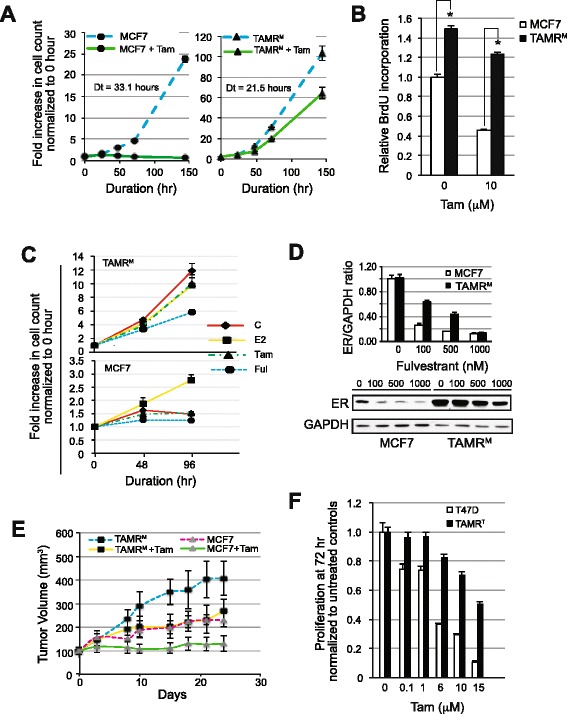
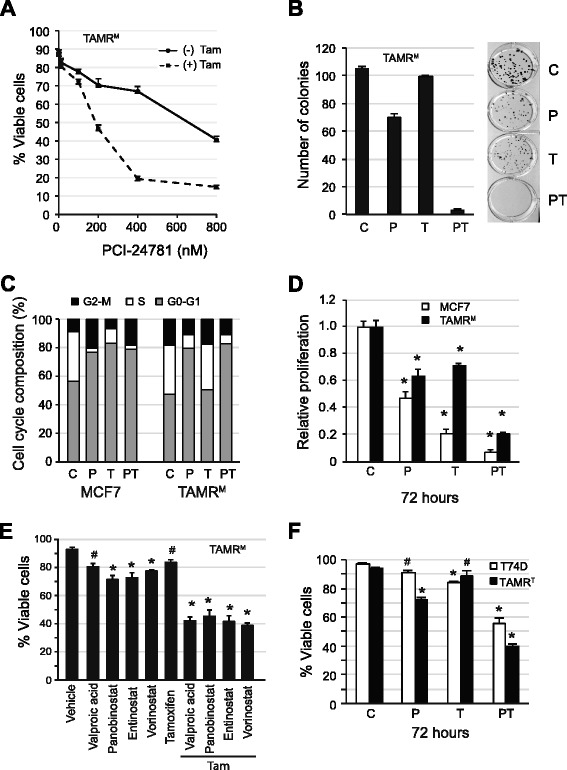
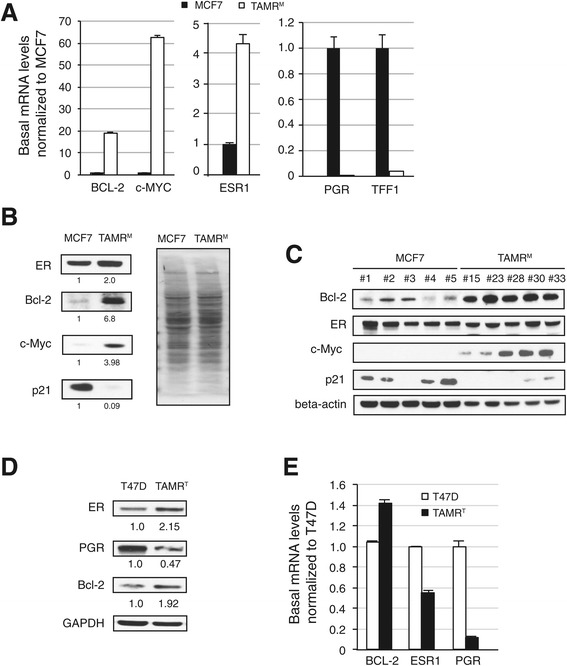
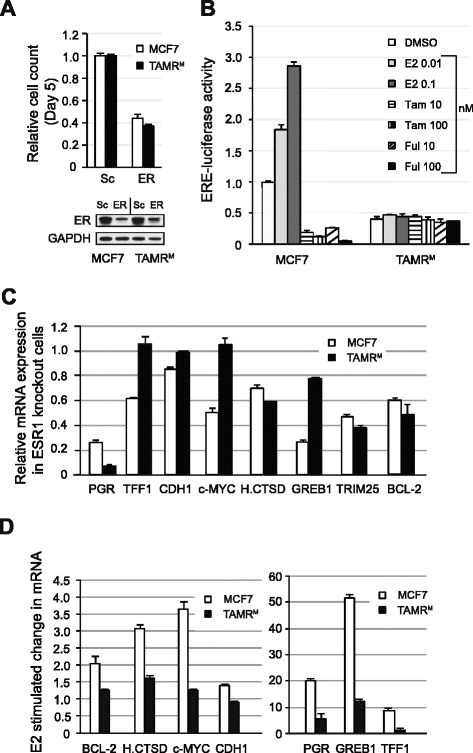
The TAMRM cells displayed decreased sensitivity to tamoxifen, fulvestrant and estrogen deprivation. Consistent with previous models, ER expression was retained and the gene harbored no mutations. Compared to parental MCF7 cells, ER expression in TAMRM was elevated, while progesterone receptor (PGR) was lost. Sensitivity of ER to ligands was greatly reduced and classic ER response genes were suppressed. This model conveyed tamoxifen resistance through transcriptional upregulation of Bcl-2 and c-Myc, and downregulation of the cell cycle checkpoint protein p21, manifesting in accelerated growth and reduced cell death. Similar to TAMRM cells, the TAMRT cell line exhibited substantially decreased tamoxifen sensitivity, increased ER and Bcl-2 expression and significantly reduced PGR expression. Treatment with HDAC inhibitors reversed the altered transcriptional events and reestablished the sensitivity of the ER to tamoxifen resulting in substantial Bcl-2 downregulation, growth arrest and apoptosis. Selective inhibition of Bcl-2 mirrored these effects in presence of an HDAC inhibitor.
Our model implicates elevated ER and Bcl-2 as key drivers of anti-estrogen resistance, which can be reversed by epigenetic modulation through HDAC inhibition.
尽管雌激素受体(ER)持续表达,但激素治疗耐药性的出现仍是治愈乳腺癌的一大挑战。近期临床研究表明,组蛋白去乙酰化酶(HDAC)抑制剂介导的表观遗传调控可逆转激素治疗耐药性。然而,关于获得性激素耐药期间ER的表观遗传调控知之甚少。我们最近的II期研究表明,HDAC抑制剂可使激素治疗耐药肿瘤对抗雌激素他莫昔芬重新敏感。在本研究中,我们试图了解这种联合治疗疗效背后的机制。
我们通过分别将ER阳性的MCF7和T47D细胞连续暴露于4-羟基他莫昔芬超过12个月,建立了对他莫昔芬耐药的细胞系,命名为TAMRM和TAMRT。使用HDAC抑制以及对包括ER和Bcl-2在内的关键生存途径进行药理学和基因操作,来表征这些耐药模型。
TAMRM细胞对他莫昔芬、氟维司群和雌激素剥夺的敏感性降低。与之前的模型一致,ER表达得以保留且该基因无突变。与亲本MCF7细胞相比,TAMRM中的ER表达升高,而孕激素受体(PGR)丢失。ER对配体的敏感性大大降低,经典的ER反应基因受到抑制。该模型通过Bcl-2和c-Myc的转录上调以及细胞周期检查点蛋白p21的下调传递他莫昔芬耐药性,表现为生长加速和细胞死亡减少。与TAMRM细胞类似,TAMRT细胞系对他莫昔芬的敏感性大幅降低,ER和Bcl-2表达增加,PGR表达显著降低。用HDAC抑制剂处理可逆转改变的转录事件,并重新建立ER对他莫昔芬的敏感性,导致Bcl-2大量下调、生长停滞和凋亡。在存在HDAC抑制剂的情况下,选择性抑制Bcl-2也呈现出类似效果。
我们的模型表明,ER和Bcl-2升高是抗雌激素耐药的关键驱动因素,可通过HDAC抑制的表观遗传调控来逆转。