Diogo Dorothée, Bastarache Lisa, Liao Katherine P, Graham Robert R, Fulton Robert S, Greenberg Jeffrey D, Eyre Steve, Bowes John, Cui Jing, Lee Annette, Pappas Dimitrios A, Kremer Joel M, Barton Anne, Coenen Marieke J H, Franke Barbara, Kiemeney Lambertus A, Mariette Xavier, Richard-Miceli Corrine, Canhão Helena, Fonseca João E, de Vries Niek, Tak Paul P, Crusius J Bart A, Nurmohamed Michael T, Kurreeman Fina, Mikuls Ted R, Okada Yukinori, Stahl Eli A, Larson David E, Deluca Tracie L, O'Laughlin Michelle, Fronick Catrina C, Fulton Lucinda L, Kosoy Roman, Ransom Michael, Bhangale Tushar R, Ortmann Ward, Cagan Andrew, Gainer Vivian, Karlson Elizabeth W, Kohane Isaac, Murphy Shawn N, Martin Javier, Zhernakova Alexandra, Klareskog Lars, Padyukov Leonid, Worthington Jane, Mardis Elaine R, Seldin Michael F, Gregersen Peter K, Behrens Timothy, Raychaudhuri Soumya, Denny Joshua C, Plenge Robert M
Division of Rheumatology, Immunology, and Allergy, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, United States of America; Division of Genetics, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, United States of America; Program in Medical and Population Genetics, Broad Institute, Cambridge, Massachusetts, United States of America; Partners HealthCare Center for Personalized Genetic Medicine, Boston, Massachusetts, United States of America.
Department of Biomedical Informatics, Vanderbilt University, Nashville, Tennessee, United States of America.
PLoS One. 2015 Apr 7;10(4):e0122271. doi: 10.1371/journal.pone.0122271. eCollection 2015.
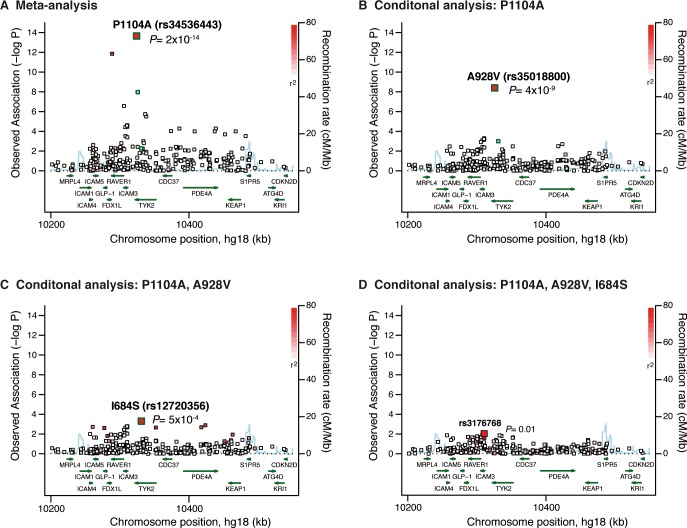
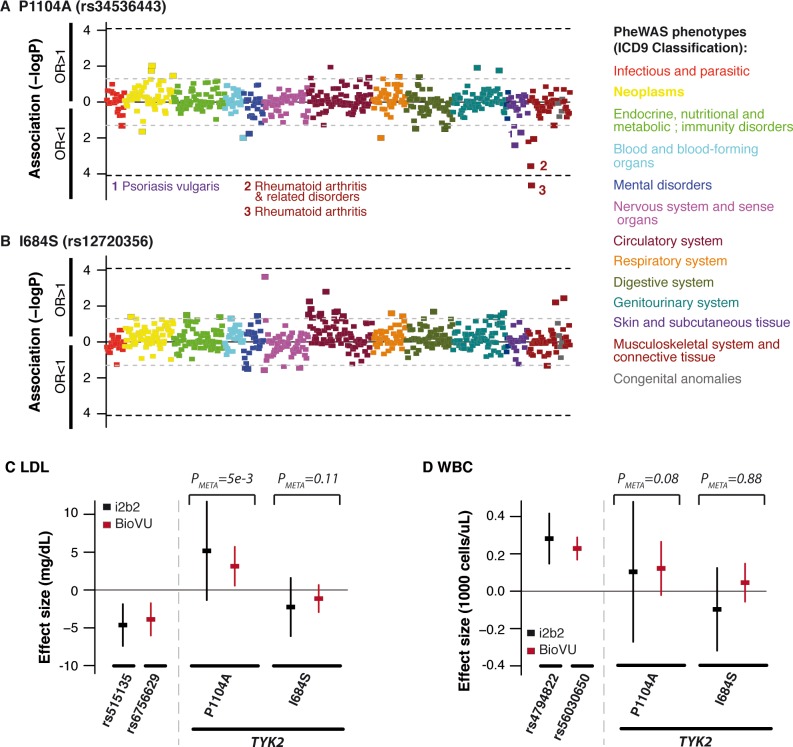
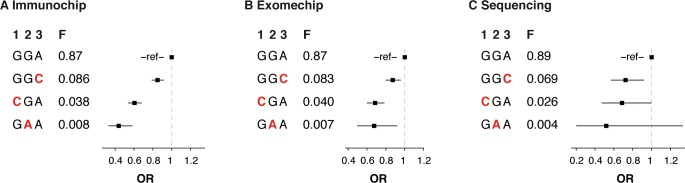
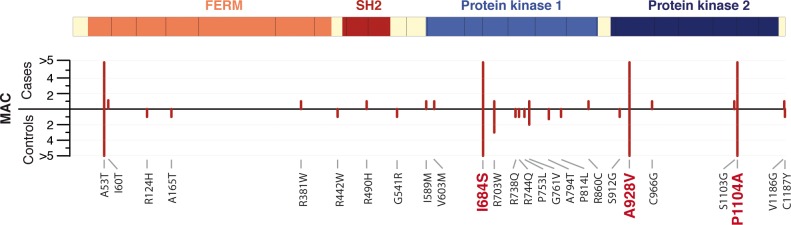
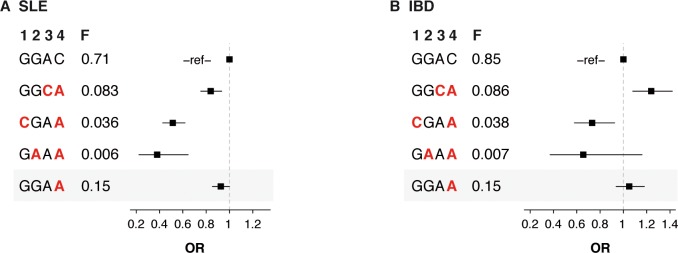
Despite the success of genome-wide association studies (GWAS) in detecting a large number of loci for complex phenotypes such as rheumatoid arthritis (RA) susceptibility, the lack of information on the causal genes leaves important challenges to interpret GWAS results in the context of the disease biology. Here, we genetically fine-map the RA risk locus at 19p13 to define causal variants, and explore the pleiotropic effects of these same variants in other complex traits. First, we combined Immunochip dense genotyping (n = 23,092 case/control samples), Exomechip genotyping (n = 18,409 case/control samples) and targeted exon-sequencing (n = 2,236 case/controls samples) to demonstrate that three protein-coding variants in TYK2 (tyrosine kinase 2) independently protect against RA: P1104A (rs34536443, OR = 0.66, P = 2.3 x 10(-21)), A928V (rs35018800, OR = 0.53, P = 1.2 x 10(-9)), and I684S (rs12720356, OR = 0.86, P = 4.6 x 10(-7)). Second, we show that the same three TYK2 variants protect against systemic lupus erythematosus (SLE, Pomnibus = 6 x 10(-18)), and provide suggestive evidence that two of the TYK2 variants (P1104A and A928V) may also protect against inflammatory bowel disease (IBD; P(omnibus) = 0.005). Finally, in a phenome-wide association study (PheWAS) assessing >500 phenotypes using electronic medical records (EMR) in >29,000 subjects, we found no convincing evidence for association of P1104A and A928V with complex phenotypes other than autoimmune diseases such as RA, SLE and IBD. Together, our results demonstrate the role of TYK2 in the pathogenesis of RA, SLE and IBD, and provide supporting evidence for TYK2 as a promising drug target for the treatment of autoimmune diseases.
尽管全基因组关联研究(GWAS)在检测类风湿性关节炎(RA)易感性等复杂表型的大量基因座方面取得了成功,但由于缺乏因果基因信息,在疾病生物学背景下解释GWAS结果面临重大挑战。在此,我们对19p13上的RA风险基因座进行遗传精细定位以确定因果变异,并探索这些相同变异在其他复杂性状中的多效性作用。首先,我们结合免疫芯片密集基因分型(n = 23,092例/对照样本)、外显子芯片基因分型(n = 18,409例/对照样本)和靶向外显子测序(n = 2,236例/对照样本),证明酪氨酸激酶2(TYK2)中的三个蛋白质编码变异独立地对RA具有保护作用:P1104A(rs34536443,OR = 0.66,P = 2.3×10⁻²¹)、A928V(rs35018800,OR = 0.53,P = 1.2×10⁻⁹)和I684S(rs12720356,OR = 0.86,P = 4.6×10⁻⁷)。其次,我们表明相同的三个TYK2变异对系统性红斑狼疮(SLE,综合P = 6×10⁻¹⁸)具有保护作用,并提供了提示性证据表明两个TYK2变异(P1104A和A928V)也可能对炎症性肠病(IBD;综合P = 0.005)具有保护作用。最后,在一项使用超过29,000名受试者的电子病历(EMR)评估>500种表型的全表型关联研究(PheWAS)中,我们没有发现令人信服的证据表明P1104A和A928V与除RA、SLE和IBD等自身免疫性疾病之外的复杂表型相关联。总之,我们的结果证明了TYK2在RA、SLE和IBD发病机制中的作用,并为TYK2作为治疗自身免疫性疾病的有前景的药物靶点提供了支持性证据。