Slobbe Paul, Windhorst Albert D, Stigter-van Walsum Marijke, Smit Egbert F, Niessen Heiko G, Solca Flavio, Stehle Gerd, van Dongen Guus A M S, Poot Alex J
Department of Radiology and Nuclear Medicine, VU University Medical Center, De Boelelaan 1117, Amsterdam, 1081 HV The Netherlands ; Department of Otolaryngology/Head and Neck Surgery, VU University Medical Center, De Boelelaan 1117, Amsterdam, 1081 HV The Netherlands.
Department of Radiology and Nuclear Medicine, VU University Medical Center, De Boelelaan 1117, Amsterdam, 1081 HV The Netherlands.
EJNMMI Res. 2015 Mar 20;5:14. doi: 10.1186/s13550-015-0088-0. eCollection 2015.
Tyrosine kinase inhibitors (TKIs) have experienced a tremendous boost in the last decade, where more than 15 small molecule TKIs have been approved by the FDA. Unfortunately, despite their promising clinical successes, a large portion of patients remain unresponsive to these targeted drugs. For non-small cell lung cancer (NSCLC), the effectiveness of TKIs is dependent on the mutational status of epidermal growth factor receptor (EGFR). The exon 19 deletion as well as the L858R point mutation lead to excellent sensitivity to TKIs such as erlotinib and gefitinib; however, despite initial good response, most patients invariably develop resistance against these first-generation reversible TKIs, e.g., via T790M point mutation. Second-generation TKIs that irreversibly bind to EGFR wild-type and mutant isoforms have therefore been developed and one of these candidates, afatinib, has now reached the market. Whether irreversible TKIs differ from reversible TKIs in their in vivo tumor-targeting properties is, however, not known and is the subject of the present study.
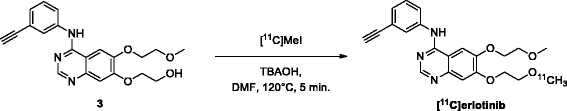
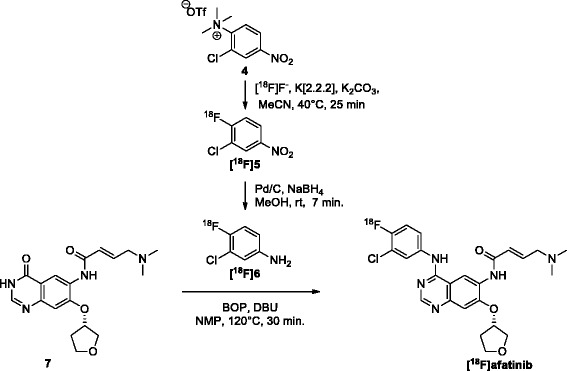
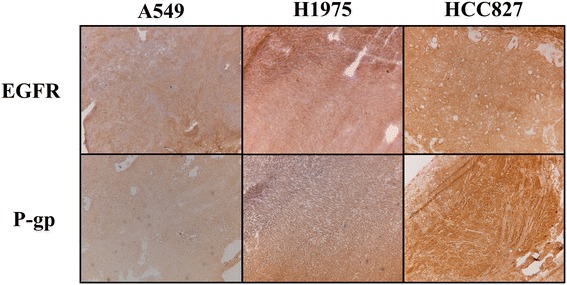
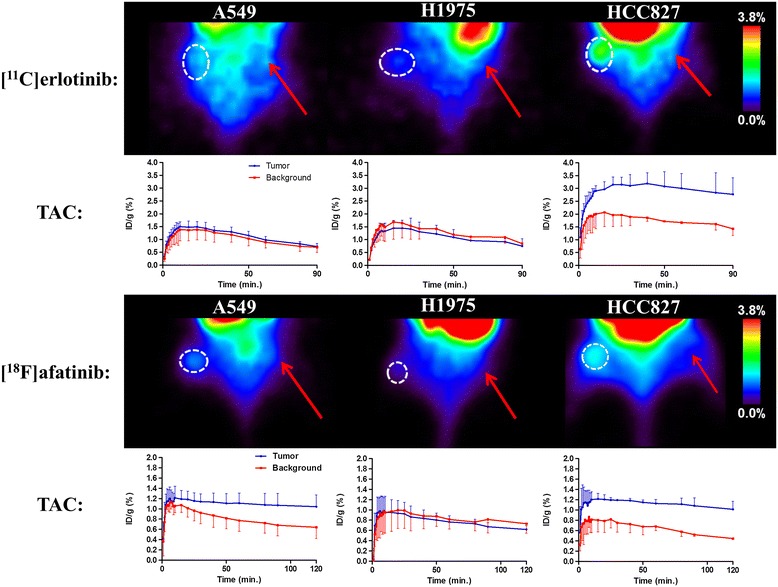
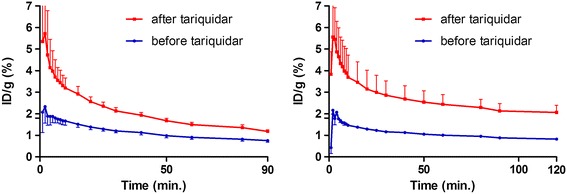
Erlotinib was labeled with carbon-11 and afatinib with fluorine-18 without modifying the structure of these compounds. A preclinical positron emission tomography (PET) study was performed in mice bearing NSCLC xenografts with a representative panel of mutations: an EGFR-WT xenograft cell line (A549), an acquired treatment-resistant L858R/T790M mutant (H1975), and a treatment-sensitive exon 19 deleted mutant (HCC827). PET imaging was performed in these xenografts with both tracers. Additionally, the effect of drug efflux transporter permeability glycoprotein (P-gp) on the tumor uptake of tracers was explored by therapeutic blocking with tariquidar.
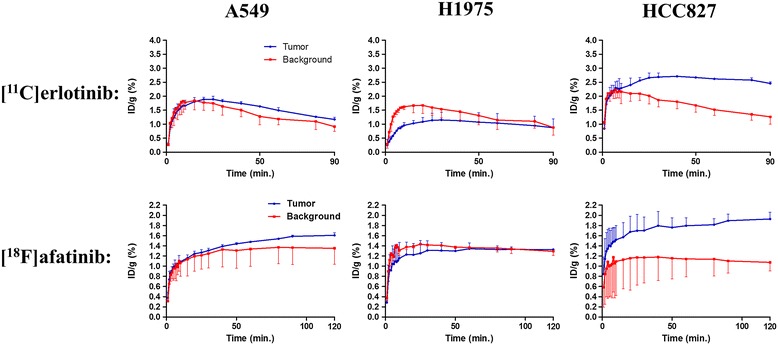
Both tracers only demonstrated selective tumor uptake in the HCC827 xenograft line (tumor-to-background ratio, [(11)C]erlotinib 1.9 ± 0.5 and [(18)F]afatinib 2.3 ± 0.4), thereby showing the ability to distinguish sensitizing mutations in vivo. No major differences were observed in the kinetics of the reversible and the irreversible tracers in each of the xenograft models. Under P-gp blocking conditions, no significant changes in tumor-to-background ratio were observed; however, [(18)F]afatinib demonstrated better tumor retention in all xenograft models.
TKI-PET provides a method to image sensitizing mutations and can be a valuable tool to compare the distinguished targeting properties of TKIs in vivo.
在过去十年中,酪氨酸激酶抑制剂(TKIs)得到了极大的发展,超过15种小分子TKIs已获美国食品药品监督管理局(FDA)批准。不幸的是,尽管它们在临床应用中取得了令人瞩目的成功,但仍有很大一部分患者对这些靶向药物无反应。对于非小细胞肺癌(NSCLC),TKIs的疗效取决于表皮生长因子受体(EGFR)的突变状态。第19外显子缺失以及L858R点突变会使患者对厄洛替尼和吉非替尼等TKIs具有极高的敏感性;然而,尽管初始反应良好,但大多数患者最终都会对这些第一代可逆性TKIs产生耐药性,例如通过T790M点突变。因此,人们开发了第二代不可逆结合EGFR野生型和突变体亚型的TKIs,其中一种候选药物阿法替尼现已上市。然而,不可逆TKIs与可逆TKIs在体内肿瘤靶向特性方面是否存在差异尚不清楚,这也是本研究的主题。
在不改变这些化合物结构的情况下,用碳-11标记厄洛替尼,用氟-18标记阿法替尼。在携带具有代表性突变组的NSCLC异种移植瘤小鼠中进行临床前正电子发射断层扫描(PET)研究:一种EGFR野生型异种移植瘤细胞系(A549)、一种获得性治疗耐药的L858R/T790M突变体(H1975)和一种对治疗敏感的第19外显子缺失突变体(HCC827)。用这两种示踪剂对这些异种移植瘤进行PET成像。此外,通过用 tariquidar进行治疗性阻断,探讨药物外排转运体通透性糖蛋白(P-gp)对示踪剂肿瘤摄取的影响。
两种示踪剂仅在HCC827异种移植瘤模型中显示出选择性肿瘤摄取(肿瘤与背景比值,[11C]厄洛替尼为1.9±0.5,[18F]阿法替尼为2.3±0.4),从而表明其具有在体内区分敏感突变的能力。在每个异种移植瘤模型中,可逆性和不可逆性示踪剂的动力学未观察到重大差异。在P-gp阻断条件下,肿瘤与背景比值未观察到显著变化;然而,[18F]阿法替尼在所有异种移植瘤模型中显示出更好的肿瘤滞留性。
TKI-PET提供了一种对敏感突变进行成像的方法,并且可以成为比较TKIs在体内独特靶向特性的有价值工具。