Mitra Abhishek, Skrzypczak Magdalena, Ginalski Krzysztof, Rowicka Maga
Department of Biochemistry and Molecular Biology, University of Texas Medical Branch at Galveston, 301 University Blvd, Galveston, TX, 77555, USA; Institute for Translational Sciences, University of Texas Medical Branch at Galveston, 301 University Blvd, Galveston, TX, 77555, USA.
Laboratory of Bioinformatics and Systems Biology, Centre of New Technologies, University of Warsaw, Zwirki i Wigury 93, 02-089 Warsaw, Poland.
PLoS One. 2015 Apr 10;10(4):e0120520. doi: 10.1371/journal.pone.0120520. eCollection 2015.
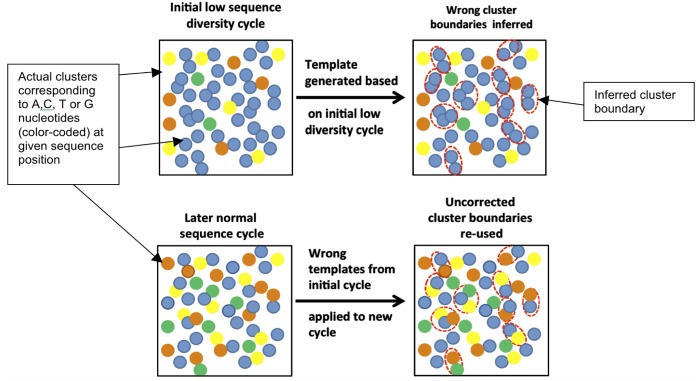
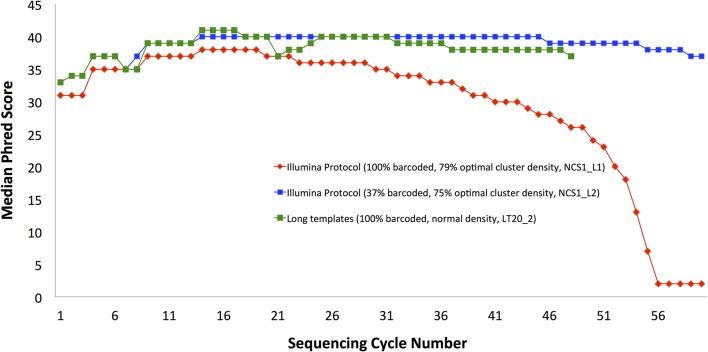
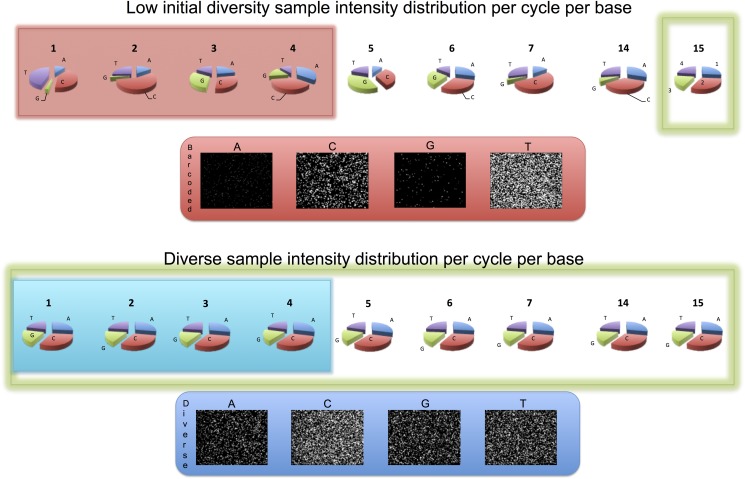
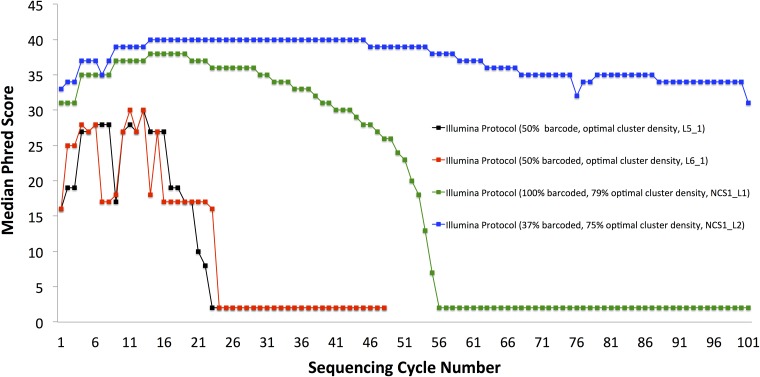
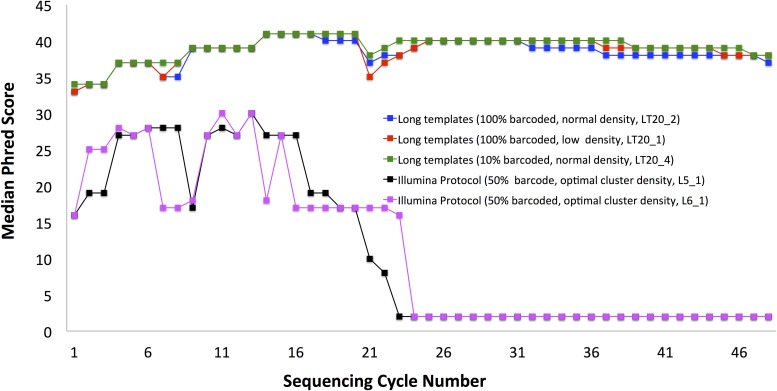
Sequencing microRNA, reduced representation sequencing, Hi-C technology and any method requiring the use of in-house barcodes result in sequencing libraries with low initial sequence diversity. Sequencing such data on the Illumina platform typically produces low quality data due to the limitations of the Illumina cluster calling algorithm. Moreover, even in the case of diverse samples, these limitations are causing substantial inaccuracies in multiplexed sample assignment (sample bleeding). Such inaccuracies are unacceptable in clinical applications, and in some other fields (e.g. detection of rare variants). Here, we discuss how both problems with quality of low-diversity samples and sample bleeding are caused by incorrect detection of clusters on the flowcell during initial sequencing cycles. We propose simple software modifications (Long Template Protocol) that overcome this problem. We present experimental results showing that our Long Template Protocol remarkably increases data quality for low diversity samples, as compared with the standard analysis protocol; it also substantially reduces sample bleeding for all samples. For comprehensiveness, we also discuss and compare experimental results from alternative approaches to sequencing low diversity samples. First, we discuss how the low diversity problem, if caused by barcodes, can be avoided altogether at the barcode design stage. Second and third, we present modified guidelines, which are more stringent than the manufacturer's, for mixing low diversity samples with diverse samples and lowering cluster density, which in our experience consistently produces high quality data from low diversity samples. Fourth and fifth, we present rescue strategies that can be applied when sequencing results in low quality data and when there is no more biological material available. In such cases, we propose that the flowcell be re-hybridized and sequenced again using our Long Template Protocol. Alternatively, we discuss how analysis can be repeated from saved sequencing images using the Long Template Protocol to increase accuracy.
对微小RNA进行测序、简化代表性测序、Hi-C技术以及任何需要使用内部条形码的方法,都会导致测序文库的初始序列多样性较低。由于Illumina簇识别算法的局限性,在Illumina平台上对这类数据进行测序通常会产生低质量的数据。此外,即使是在样本多样的情况下,这些局限性也会在多重样本分配(样本串扰)中导致大量不准确的情况。这种不准确在临床应用以及其他一些领域(例如罕见变异的检测)中是不可接受的。在此,我们讨论了低多样性样本质量问题和样本串扰这两个问题是如何在初始测序循环期间由流动池上簇的错误检测所导致的。我们提出了简单的软件修改方法(长模板协议)来克服这个问题。我们展示的实验结果表明,与标准分析协议相比,我们的长模板协议显著提高了低多样性样本的数据质量;它还大幅减少了所有样本的样本串扰。为了全面性,我们还讨论并比较了对低多样性样本进行测序的替代方法的实验结果。首先,我们讨论如果低多样性问题是由条形码引起的,如何在条形码设计阶段完全避免这个问题。第二和第三,我们给出了比制造商的指南更严格的修改后的指南,用于将低多样性样本与多样样本混合以及降低簇密度,根据我们的经验,这始终能从低多样性样本中产生高质量的数据。第四和第五,我们提出了在测序产生低质量数据且没有更多生物材料可用时可以应用的挽救策略。在这种情况下,我们建议使用我们的长模板协议对流动池进行重新杂交并再次测序。或者,我们讨论如何使用长模板协议从保存的测序图像中重复进行分析以提高准确性。