Williams Jeffrey M, Inoue Takamasa, Chen Grace, Tsai Billy
Department of Cell and Developmental Biology, University of Michigan Medical School, Ann Arbor, MI 48103.
Department of Cell and Developmental Biology, University of Michigan Medical School, Ann Arbor, MI 48103
Mol Biol Cell. 2015 Jun 15;26(12):2181-9. doi: 10.1091/mbc.E15-01-0014. Epub 2015 Apr 15.
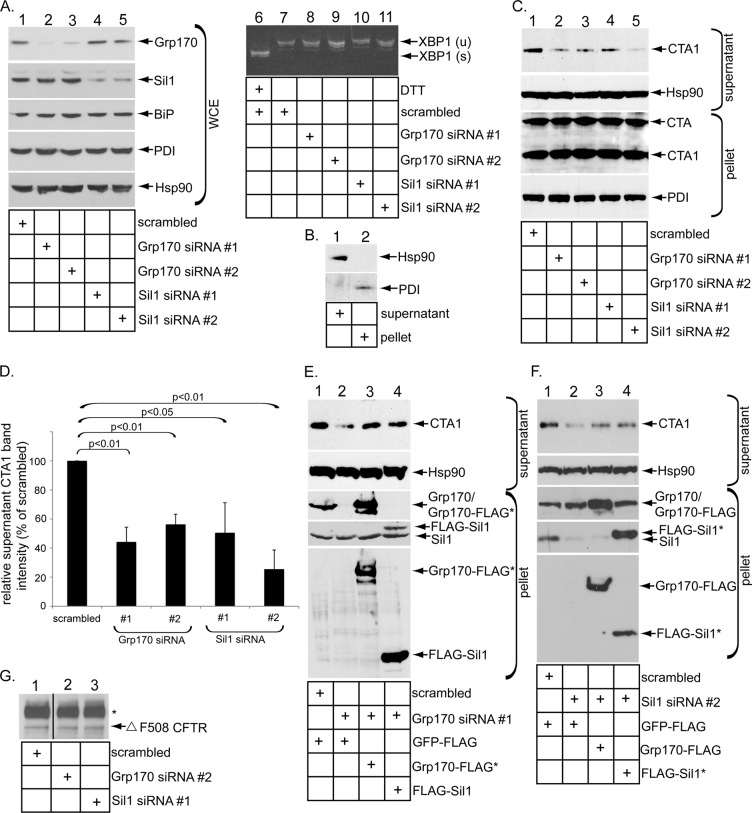
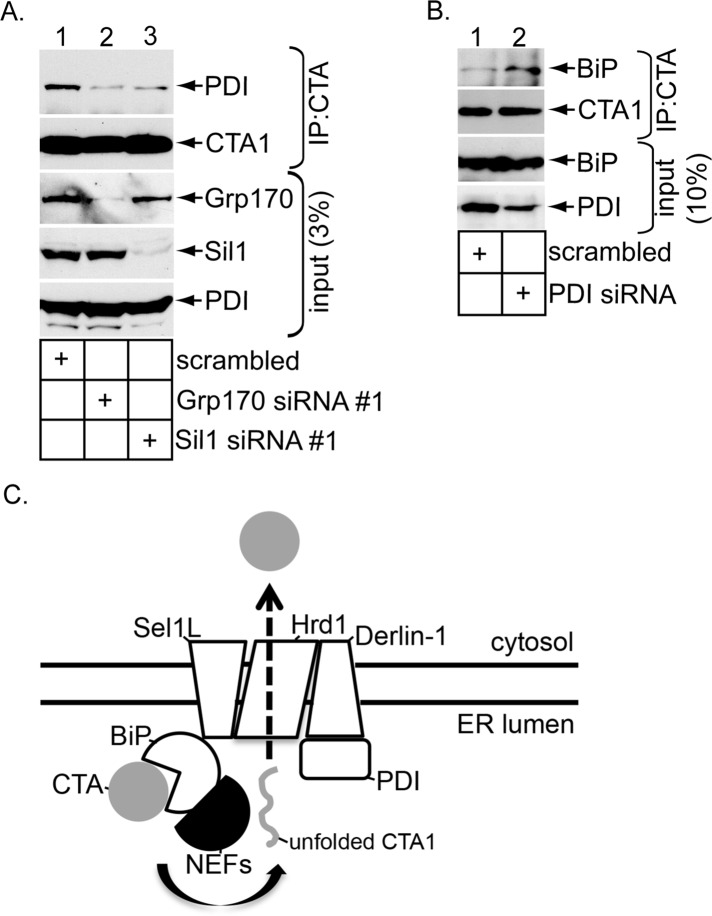
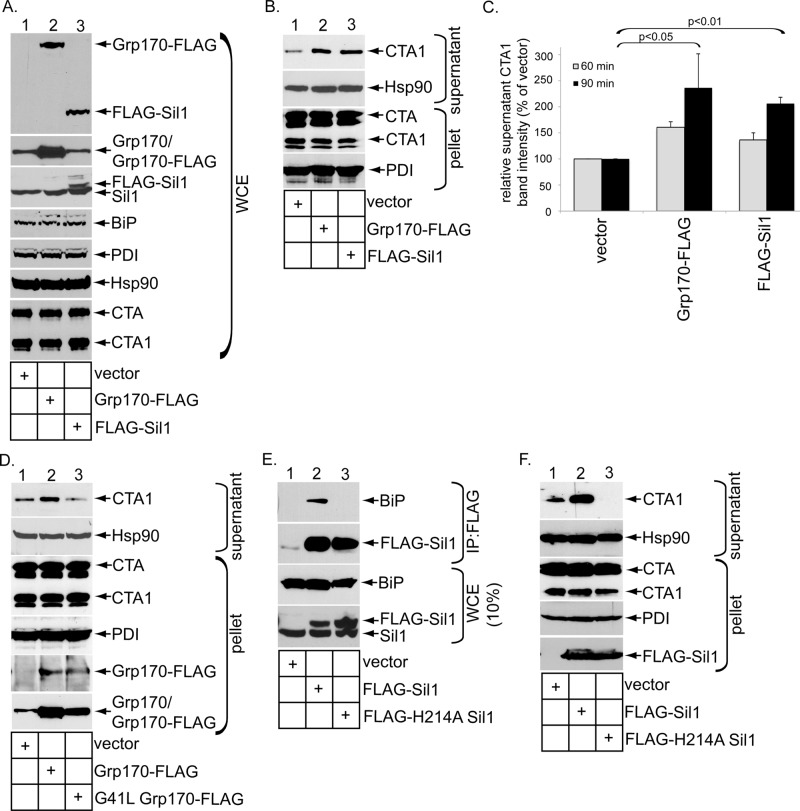
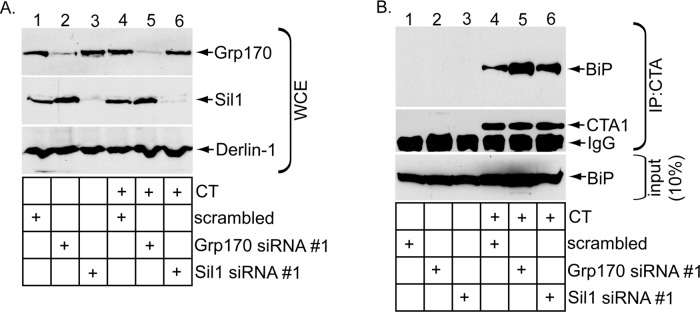
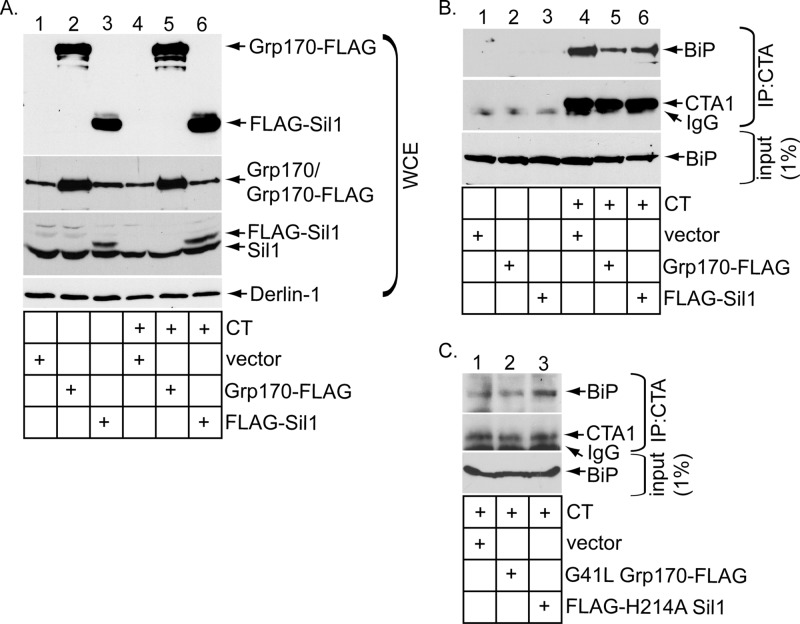
Cholera toxin (CT) intoxicates cells by trafficking from the cell surface to the endoplasmic reticulum (ER), where the catalytic CTA1 subunit hijacks components of the ER-associated degradation (ERAD) machinery to retrotranslocate to the cytosol and induce toxicity. In the ER, CT targets to the ERAD machinery composed of the E3 ubiquitin ligase Hrd1-Sel1L complex, in part via the activity of the Sel1L-binding partner ERdj5. This J protein stimulates BiP's ATPase activity, allowing BiP to capture the toxin. Presumably, toxin release from BiP must occur before retrotranslocation. Here, using loss-and gain-of-function approaches coupled with binding studies, we demonstrate that the ER-resident nucleotide exchange factors (NEFs) Grp170 and Sil1 induce CT release from BiP in order to promote toxin retrotranslocation. In addition, we find that after NEF-dependent release from BiP, the toxin is transferred to protein disulfide isomerase; this ER redox chaperone is known to unfold CTA1, which allows the toxin to cross the Hrd1-Sel1L complex. Our data thus identify two NEFs that trigger toxin release from BiP to enable successful retrotranslocation and clarify the fate of the toxin after it disengages from BiP.
霍乱毒素(CT)通过从细胞表面运输至内质网(ER)来使细胞中毒,在那里,催化性CTA1亚基劫持内质网相关降解(ERAD)机制的组分,以逆向转运至胞质溶胶并诱导毒性。在ER中,CT部分通过Sel1L结合伴侣ERdj5的活性靶向由E3泛素连接酶Hrd1-Sel1L复合物组成的ERAD机制。这种J蛋白刺激BiP的ATP酶活性,使BiP捕获毒素。据推测,毒素从BiP释放必定发生在逆向转运之前。在此,我们使用功能丧失和功能获得方法并结合结合研究,证明内质网驻留核苷酸交换因子(NEF)Grp170和Sil1诱导CT从BiP释放,以促进毒素逆向转运。此外,我们发现,在毒素从BiP发生NEF依赖性释放后,它会转移至蛋白二硫键异构酶;这种内质网氧化还原伴侣已知会使CTA1解折叠,从而使毒素能够穿过Hrd1-Sel1L复合物。因此,我们的数据鉴定出两种触发毒素从BiP释放以实现成功逆向转运的NEF,并阐明了毒素从BiP脱离后的去向。