Koch Johannes, Freisinger Peter, Feichtinger René G, Zimmermann Franz A, Rauscher Christian, Wagentristl Hans P, Konstantopoulou Vassiliki, Seidl Rainer, Haack Tobias B, Prokisch Holger, Ahting Uwe, Sperl Wolfgang, Mayr Johannes A, Maier Esther M
Department of Pediatrics, Paracelsus Medical University Salzburg, Muellner Hauptstr. 48, 5020, Salzburg, Austria.
Department of Pediatrics Kreisklinken Reutlingen, Steinenbergstr. 31, 72764, Reutlingen, Germany.
Orphanet J Rare Dis. 2015 Apr 2;10:40. doi: 10.1186/s13023-015-0254-5.
TTC19 deficiency is a progressive neurodegenerative disease associated with isolated mitochondrial respiratory chain (MRC) complex III deficiency and loss-of-function mutations in the TT19 gene in the few patients reported so far.
We performed exome sequencing and selective mutational analysis of TTC19, respectively, in patients from three unrelated families presenting with initially unspecific clinical signs of muscular hypotonia and global developmental delay followed by regression, ataxia, loss of speech, and rapid neurological deterioration. One patient showed severe lactic acidosis at the neonatal age and during intercurrent illness.
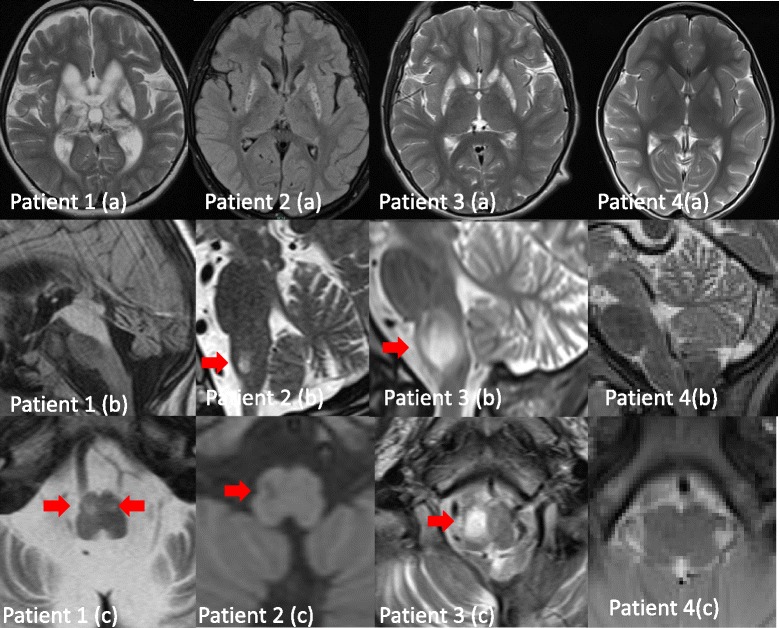
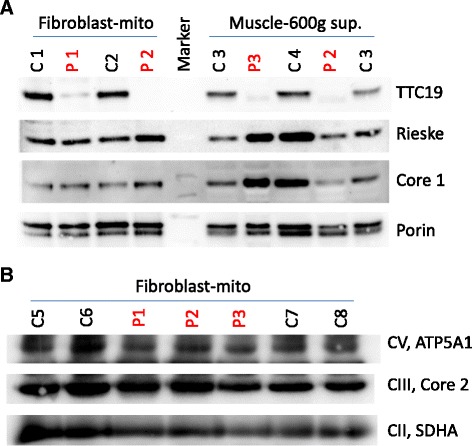
We identified homozygous mutations in all three index cases, in two families novel missense mutations (c.544 T > C/p.Leu185Pro; c.917 T > C/p.Leu324Pro). The younger sister of the severely affected patient 3 showed only mild delay of motor skills and muscular hypotonia so far but is also homozygous for the same mutation. Notably, one patient revealed normal activities of MRC complex III in two independent muscle biopsies. Neuroimaging of the severely affected patients demonstrated lesions in putamen and caudate nuclei, cerebellar atrophy, and the unusual finding of hypertrophic olivary nuclei degeneration. Reviewing the literature revealed striking similarities regarding neuroimaging and clinical course in pediatric patients with TTC19 deficiency: patterns consistent with Leigh or Leigh-like syndrome were found in almost all, hypertrophic olivary nucleus degeneration in all patients reported so far. The clinical course in pediatric patients is characterized by an initially unspecific developmental delay, followed by regression, progressive signs and symptoms of cerebellar, basal ganglia and brainstem affection, especially loss of speech and ataxia. Subsequently, neurological deterioration leading to a vegetative state occurs.
Our findings add to the phenotypic, genetic, and biochemical spectrum of TTC19 deficiency. However, TTC19 deficient patients do show characteristic clinical and neuroimaging features, which may facilitate diagnosis of this yet rare disorder. Normal MRC complex III activity does not exclude the diagnosis.
TTC19缺乏症是一种进行性神经退行性疾病,在迄今为止报道的少数患者中,与孤立的线粒体呼吸链(MRC)复合物III缺乏症以及TT19基因的功能丧失突变有关。
我们分别对来自三个无关家庭的患者进行了外显子组测序和TTC19的选择性突变分析,这些患者最初表现为肌张力低下和全面发育迟缓等非特异性临床症状,随后出现发育倒退、共济失调、言语丧失和快速的神经功能恶化。一名患者在新生儿期和并发疾病期间出现严重乳酸酸中毒。
我们在所有三名索引病例中均鉴定出纯合突变,在两个家庭中发现了新的错义突变(c.544 T>C/p.Leu185Pro;c.917 T>C/p.Leu324Pro)。严重受影响患者3的妹妹目前仅表现出轻度运动技能延迟和肌张力低下,但也为相同突变的纯合子。值得注意的是,一名患者在两次独立的肌肉活检中显示MRC复合物III活性正常。严重受影响患者的神经影像学检查显示壳核和尾状核有病变、小脑萎缩,以及肥大性橄榄核变性这一不寻常发现。文献回顾显示,TTC19缺乏症儿科患者在神经影像学和临床病程方面存在显著相似之处:几乎所有患者都有与Leigh或Leigh样综合征一致的模式,迄今为止报道的所有患者均有肥大性橄榄核变性。儿科患者的临床病程特征为最初非特异性的发育延迟,随后出现发育倒退、小脑、基底神经节和脑干受累的进行性体征和症状,尤其是言语丧失和共济失调。随后,神经功能恶化导致植物人状态。
我们的发现增加了TTC19缺乏症的表型特征、遗传特征和生化特征。然而,TTC19缺乏症患者确实表现出特征性的临床和神经影像学特征,这可能有助于诊断这种罕见疾病。MRC复合物III活性正常并不能排除诊断。