Banach Mateusz, Prudhomme Nicolas, Carpentier Mathilde, Duprat Elodie, Papandreou Nikolaos, Kalinowska Barbara, Chomilier Jacques, Roterman Irena
Department of Bioinformatics and Telemedicine, Medical College, Jagiellonian University, Krakow, Poland.
Protein Structure Prediction group, IMPMC, UPMC & CNRS, Paris, France.
PLoS One. 2015 Apr 27;10(4):e0125098. doi: 10.1371/journal.pone.0125098. eCollection 2015.
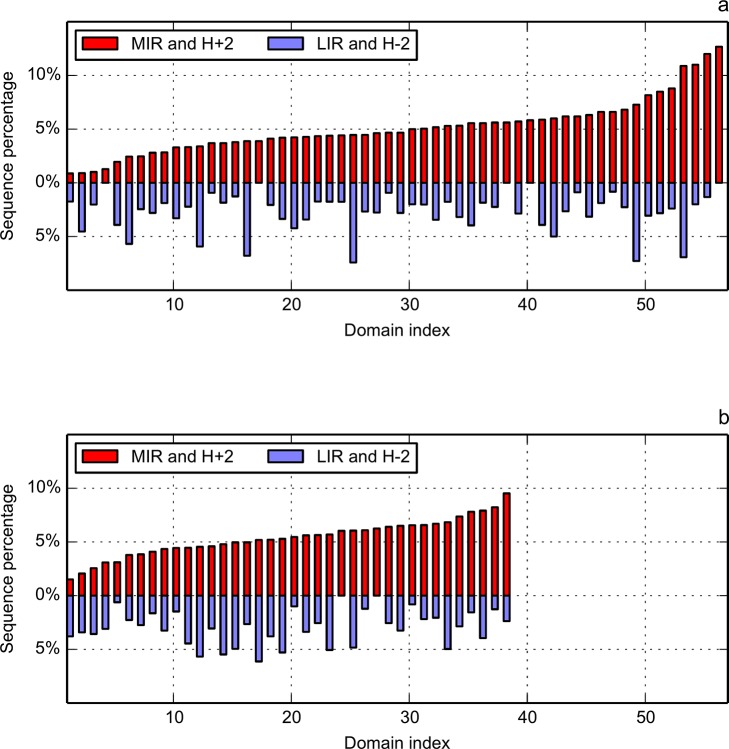
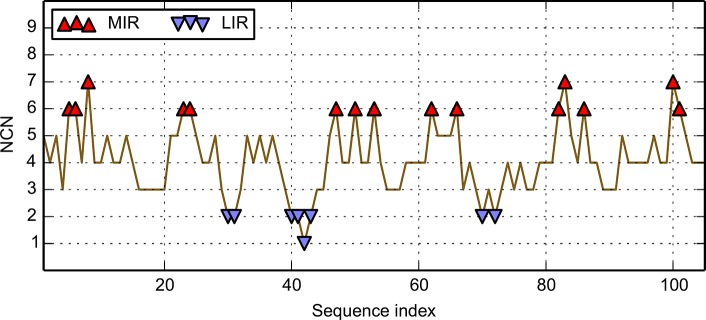
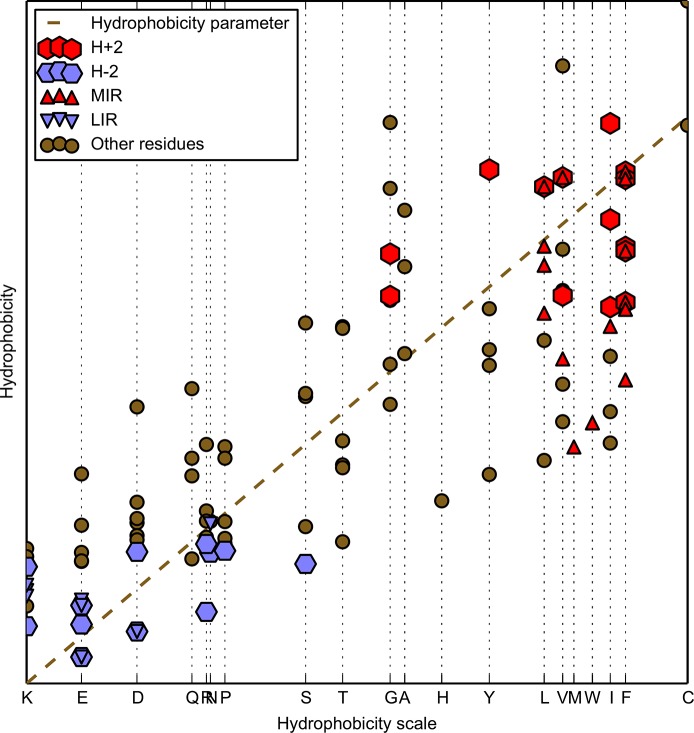
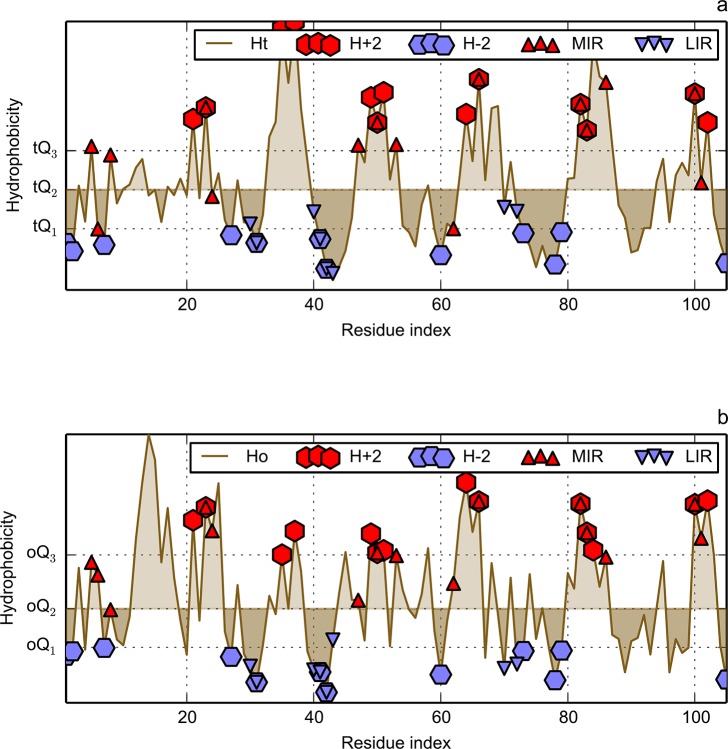
Folding nucleus of globular proteins formation starts by the mutual interaction of a group of hydrophobic amino acids whose close contacts allow subsequent formation and stability of the 3D structure. These early steps can be predicted by simulation of the folding process through a Monte Carlo (MC) coarse grain model in a discrete space. We previously defined MIRs (Most Interacting Residues), as the set of residues presenting a large number of non-covalent neighbour interactions during such simulation. MIRs are good candidates to define the minimal number of residues giving rise to a given fold instead of another one, although their proportion is rather high, typically [15-20]% of the sequences. Having in mind experiments with two sequences of very high levels of sequence identity (up to 90%) but different folds, we combined the MIR method, which takes sequence as single input, with the "fuzzy oil drop" (FOD) model that requires a 3D structure, in order to estimate the residues coding for the fold. FOD assumes that a globular protein follows an idealised 3D Gaussian distribution of hydrophobicity density, with the maximum in the centre and minima at the surface of the "drop". If the actual local density of hydrophobicity around a given amino acid is as high as the ideal one, then this amino acid is assigned to the core of the globular protein, and it is assumed to follow the FOD model. Therefore one obtains a distribution of the amino acids of a protein according to their agreement or rejection with the FOD model.
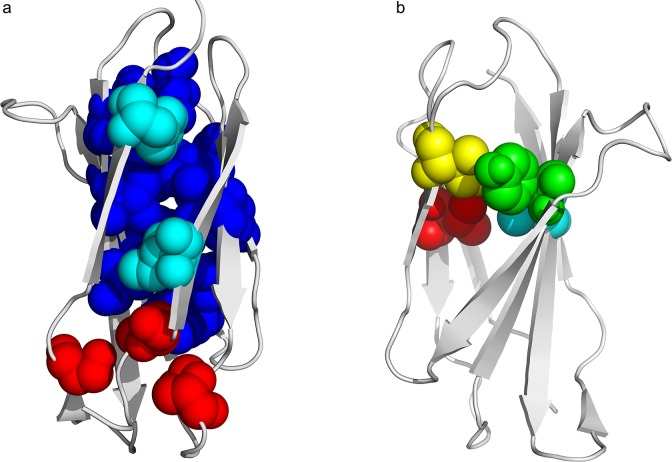
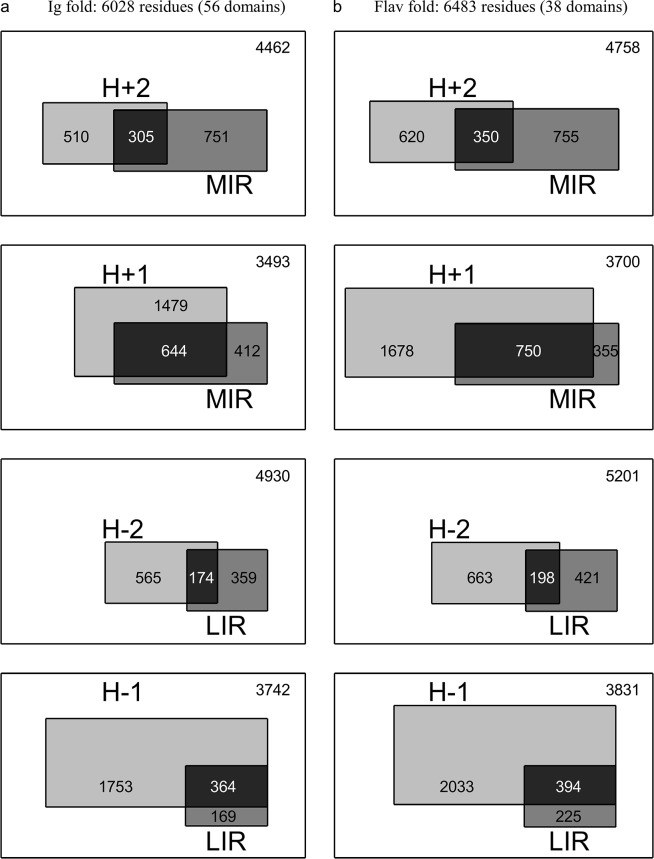
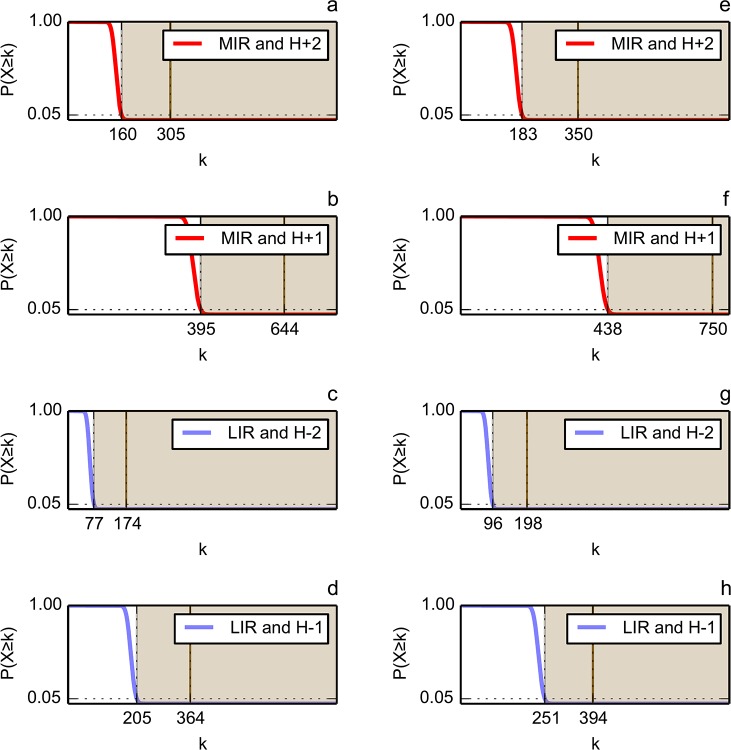
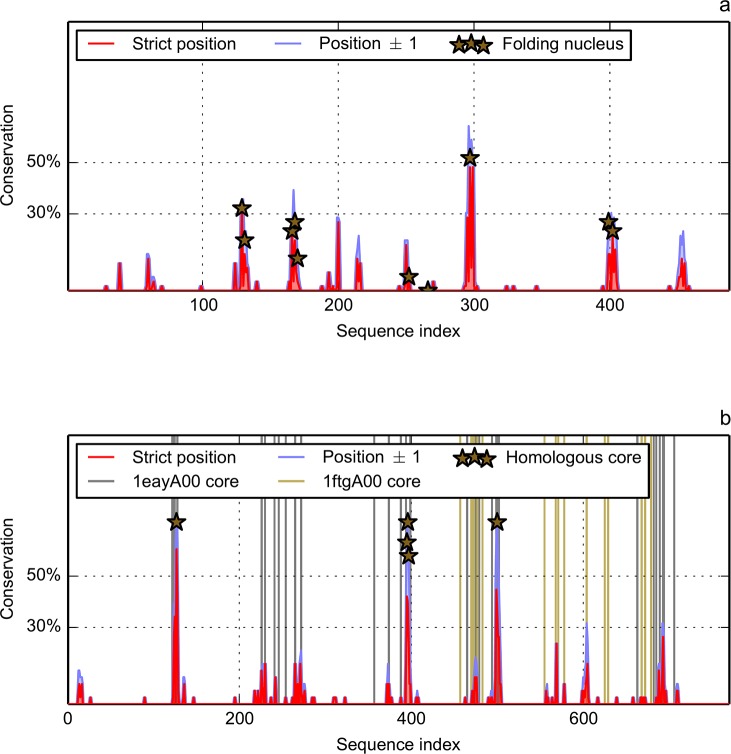
We compared and combined MIR and FOD methods to define the minimal nucleus, or keystone, of two populated folds: immunoglobulin-like (Ig) and flavodoxins (Flav). The combination of these two approaches defines some positions both predicted as a MIR and assigned as accordant with the FOD model. It is shown here that for these two folds, the intersection of the predicted sets of residues significantly differs from random selection. It reduces the number of selected residues by each individual method and allows a reasonable agreement with experimentally determined key residues coding for the particular fold. In addition, the intersection of the two methods significantly increases the specificity of the prediction, providing a robust set of residues that constitute the folding nucleus.
球状蛋白质折叠核的形成始于一组疏水氨基酸的相互作用,这些氨基酸的紧密接触使得后续三维结构的形成和稳定性得以实现。这些早期步骤可以通过在离散空间中通过蒙特卡罗(MC)粗粒模型模拟折叠过程来预测。我们之前定义了最相互作用残基(MIRs),即在此类模拟过程中呈现大量非共价邻域相互作用的残基集合。MIRs是定义产生特定折叠而非其他折叠所需最小残基数的良好候选者,尽管它们的比例相当高,通常占序列的[15 - 20]%。考虑到对两个具有非常高序列同一性水平(高达90%)但折叠不同的序列进行的实验,我们将以序列作为单一输入的MIR方法与需要三维结构的“模糊油滴”(FOD)模型相结合,以估计编码折叠的残基。FOD假设球状蛋白质遵循理想化的疏水性密度三维高斯分布,在“油滴”中心最大,表面最小。如果给定氨基酸周围的实际局部疏水性密度与理想密度一样高,那么该氨基酸就被分配到球状蛋白质的核心,并假定其遵循FOD模型。因此,根据蛋白质氨基酸与FOD模型的符合或不符合情况,可以得到蛋白质氨基酸的一种分布。
我们比较并结合了MIR和FOD方法来定义两种常见折叠的最小核或关键部分:免疫球蛋白样(Ig)和黄素氧还蛋白(Flav)。这两种方法的结合确定了一些既被预测为MIR又被指定为符合FOD模型的位置。此处表明,对于这两种折叠,预测的残基集的交集与随机选择有显著差异。它减少了每种单独方法选择的残基数量,并与实验确定的编码特定折叠的关键残基有合理的一致性。此外,两种方法的交集显著提高了预测的特异性,提供了一组构成折叠核的稳健残基。