Gibbons Jacqueline A, de Vries Michiel, Krauwinkel Walter, Ohtsu Yoshiaki, Noukens Jan, van der Walt Jan-Stefan, Mol Roelof, Mordenti Joyce, Ouatas Taoufik
Medivation, Inc., 525 Market Street, 36th Floor, San Francisco, CA, 94105, USA.
Astellas Pharma Europe B.V., Leiden, The Netherlands.
Clin Pharmacokinet. 2015 Oct;54(10):1057-69. doi: 10.1007/s40262-015-0283-1.
Two phase I drug interaction studies were performed with oral enzalutamide, which is approved for the treatment of metastatic castration-resistant prostate cancer (mCRPC).
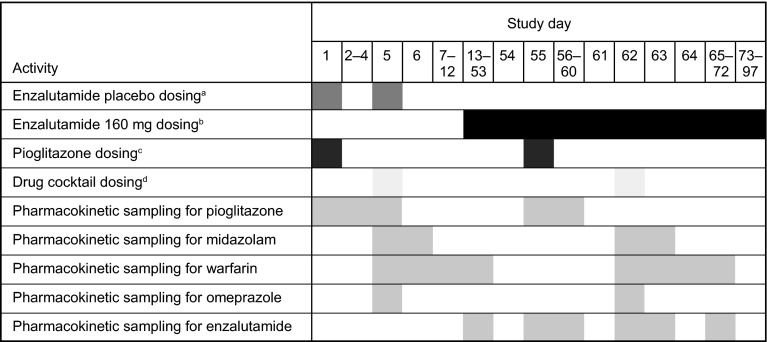
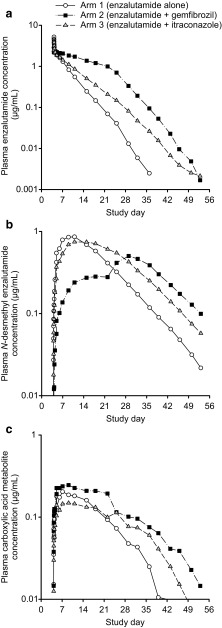
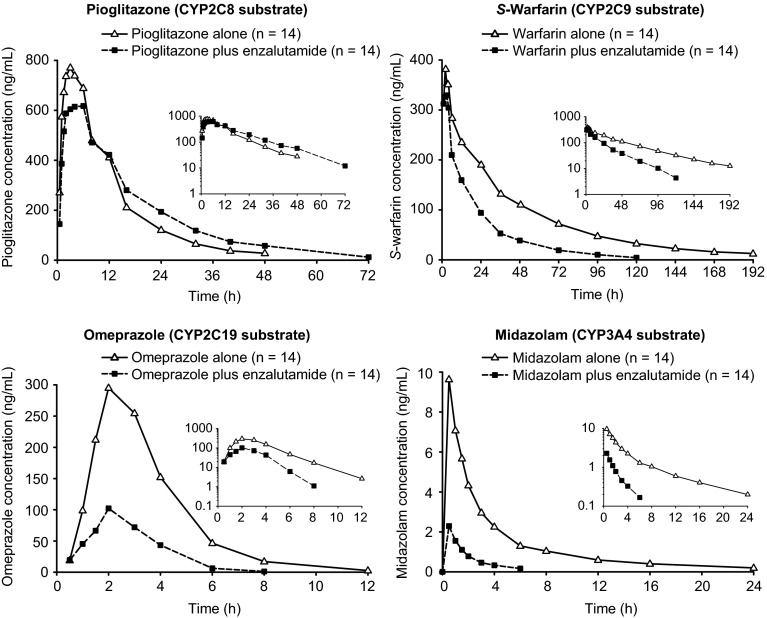
A parallel-treatment design (n = 41) was used to evaluate the effects of a strong cytochrome P450 (CYP) 2C8 inhibitor (oral gemfibrozil 600 mg twice daily) or strong CYP3A4 inhibitor (oral itraconazole 200 mg once daily) on the pharmacokinetics of enzalutamide and its active metabolite N-desmethyl enzalutamide after a single dose of enzalutamide (160 mg). A single-sequence crossover design (n = 14) was used to determine the effects of enzalutamide 160 mg/day on the pharmacokinetics of a single oral dose of sensitive substrates for CYP2C8 (pioglitazone 30 mg), CYP2C9 (warfarin 10 mg), CYP2C19 (omeprazole 20 mg), or CYP3A4 (midazolam 2 mg).
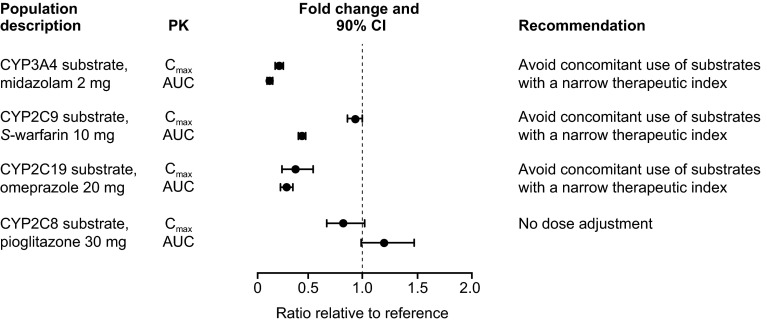
Coadministration of gemfibrozil increased the composite area under the plasma concentration-time curve from time zero to infinity (AUC∞) of enzalutamide plus active metabolite by 2.2-fold, and coadministration of itraconazole increased the composite AUC∞ by 1.3-fold. Enzalutamide did not affect exposure to oral pioglitazone. Enzalutamide reduced the AUC∞ of oral S-warfarin, omeprazole, and midazolam by 56, 70, and 86 %, respectively; therefore, enzalutamide is a moderate inducer of CYP2C9 and CYP2C19 and a strong inducer of CYP3A4.
If a patient requires coadministration of a strong CYP2C8 inhibitor with enzalutamide, then the enzalutamide dose should be reduced to 80 mg/day. It is recommended to avoid concomitant use of enzalutamide with narrow therapeutic index drugs metabolized by CYP2C9, CYP2C19, or CYP3A4, as enzalutamide may decrease their exposure.
开展了两项I期药物相互作用研究,涉及已获批准用于治疗转移性去势抵抗性前列腺癌(mCRPC)的口服恩杂鲁胺。
采用平行治疗设计(n = 41)评估强效细胞色素P450(CYP)2C8抑制剂(口服吉非贝齐600 mg,每日两次)或强效CYP3A4抑制剂(口服伊曲康唑200 mg,每日一次)对单剂量恩杂鲁胺(160 mg)后恩杂鲁胺及其活性代谢物N-去甲基恩杂鲁胺药代动力学的影响。采用单序列交叉设计(n = 14)确定每日160 mg恩杂鲁胺对单剂量口服CYP2C8敏感底物(吡格列酮30 mg)、CYP2C9(华法林10 mg)、CYP2C19(奥美拉唑20 mg)或CYP3A4(咪达唑仑2 mg)药代动力学的影响。
吉非贝齐与恩杂鲁胺合用使恩杂鲁胺加活性代谢物从时间零点至无穷大的血浆浓度-时间曲线下面积(AUC∞)增加2.2倍,伊曲康唑与恩杂鲁胺合用使复合AUC∞增加1.3倍。恩杂鲁胺不影响口服吡格列酮的暴露量。恩杂鲁胺使口服S-华法林、奥美拉唑和咪达唑仑的AUC∞分别降低56%、70%和86%;因此,恩杂鲁胺是CYP2C9和CYP2C19的中度诱导剂以及CYP3A4的强效诱导剂。
如果患者需要将强效CYP2C8抑制剂与恩杂鲁胺合用,则恩杂鲁胺剂量应减至80 mg/天。建议避免恩杂鲁胺与经CYP2C9、CYP2C19或CYP3A4代谢的治疗指数窄的药物合用,因为恩杂鲁胺可能会降低它们的暴露量。