Wintermans Bastiaan, Brandt Bernd, Vandenbroucke-Grauls Christina, Budding Andries
Department of Medical Microbiology and Infection Control, VU University Medical Center, Amsterdam, The Netherlands.
Department of Preventive Dentistry, Academic Centre for Dentistry Amsterdam (ACTA), University of Amsterdam and VU University Amsterdam, Amsterdam, The Netherlands.
PLoS One. 2015 May 1;10(5):e0123851. doi: 10.1371/journal.pone.0123851. eCollection 2015.
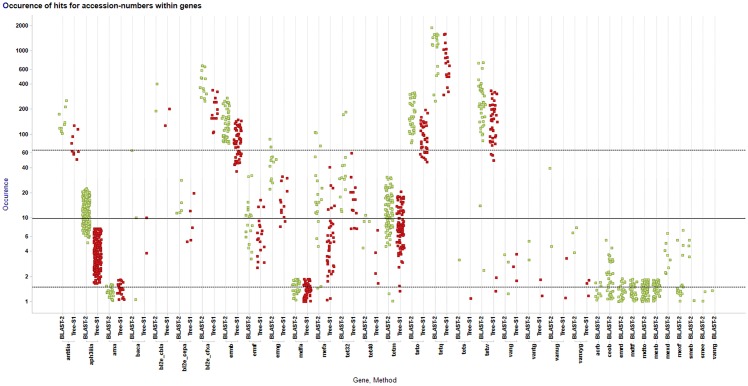
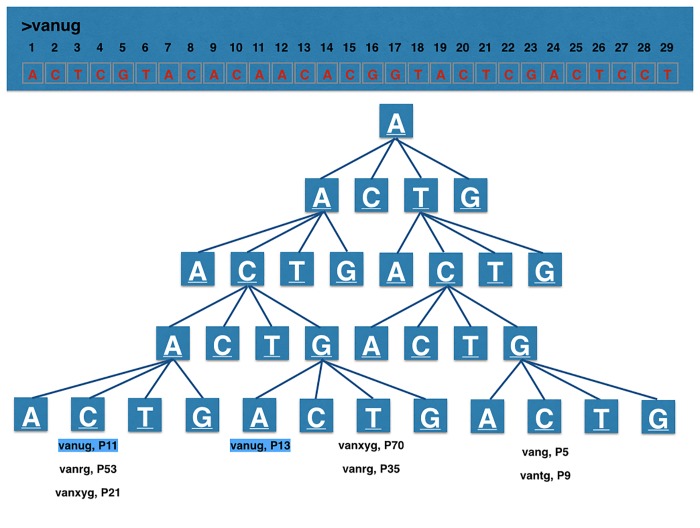
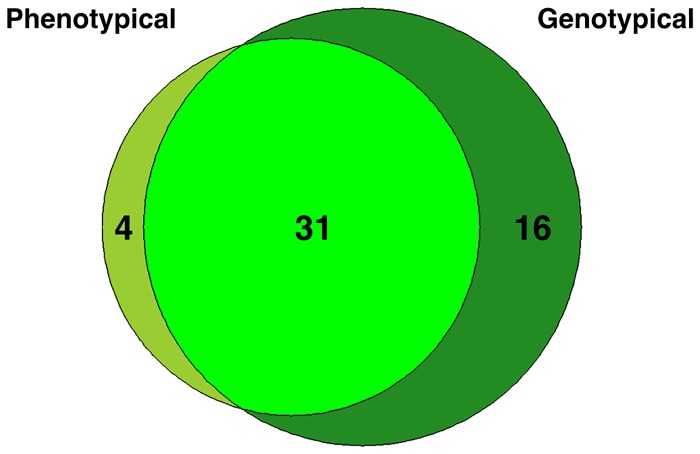
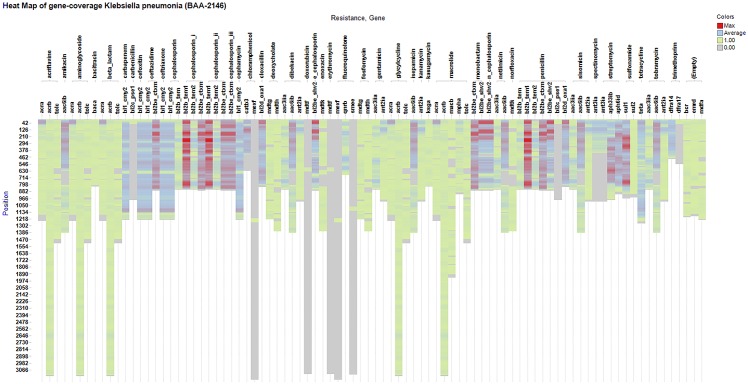
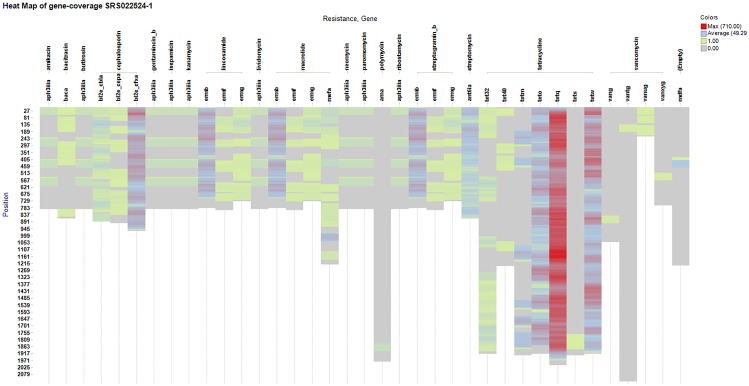
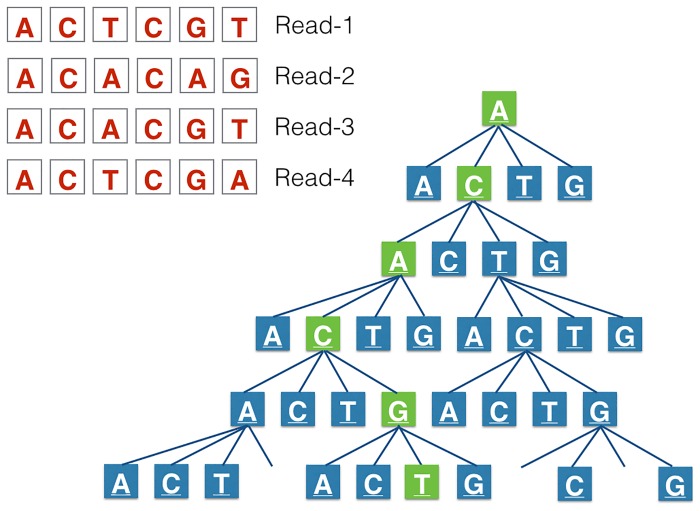
Next-generation sequencing is not yet commonly used in clinical laboratories because of a lack of simple and intuitive tools. We developed a software tool (TreeSeq) with a quaternary tree search structure for the analysis of sequence data. This permits rapid searches for sequences of interest in large datasets. We used TreeSeq to screen a gut microbiota metagenomic dataset and a whole genome sequencing (WGS) dataset of a strain of Klebsiella pneumoniae for antibiotic resistance genes and compared the results with BLAST and phenotypic resistance determination. TreeSeq was more than thirty times faster than BLAST and accurately detected resistance gene sequences in complex metagenomic data and resistance genes corresponding with the phenotypic resistance pattern of the Klebsiella strain. Resistance genes found by TreeSeq were visualized as a gene coverage heat map, aiding in the interpretation of results. TreeSeq brings analysis of metagenomic and WGS data within reach of clinical diagnostics.
由于缺乏简单直观的工具,下一代测序在临床实验室中尚未普遍使用。我们开发了一种具有四叉树搜索结构的软件工具(TreeSeq)用于序列数据分析。这使得能够在大型数据集中快速搜索感兴趣的序列。我们使用TreeSeq筛选肠道微生物群宏基因组数据集和一株肺炎克雷伯菌的全基因组测序(WGS)数据集以查找抗生素抗性基因,并将结果与BLAST和表型抗性测定进行比较。TreeSeq比BLAST快三十多倍,并且能够在复杂的宏基因组数据中准确检测抗性基因序列以及与肺炎克雷伯菌菌株表型抗性模式相对应的抗性基因。通过TreeSeq发现的抗性基因被可视化为基因覆盖热图,有助于结果的解读。TreeSeq使宏基因组和WGS数据的分析能够应用于临床诊断。