Codina-Solà Marta, Rodríguez-Santiago Benjamín, Homs Aïda, Santoyo Javier, Rigau Maria, Aznar-Laín Gemma, Del Campo Miguel, Gener Blanca, Gabau Elisabeth, Botella María Pilar, Gutiérrez-Arumí Armand, Antiñolo Guillermo, Pérez-Jurado Luis Alberto, Cuscó Ivon
Department of Experimental and Health Sciences, Universitat Pompeu Fabra, C/Doctor Aiguader 88, 422, Barcelona, 08003 Spain ; Hospital del Mar Research Institute (IMIM), C/Doctor Aiguader 88, Barcelona, 08003 Spain ; Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBER-ER), C/ Monforte de Lemos 3-5, Madrid, 28029 Spain.
Quantitative Genomic Medicine Laboratories (qGenomics), C/Doctor Aiguader 88, 422, Barcelona, 08003 Spain.
Mol Autism. 2015 Apr 15;6:21. doi: 10.1186/s13229-015-0017-0. eCollection 2015.
Autism spectrum disorders (ASD) are a group of neurodevelopmental disorders with high heritability. Recent findings support a highly heterogeneous and complex genetic etiology including rare de novo and inherited mutations or chromosomal rearrangements as well as double or multiple hits.
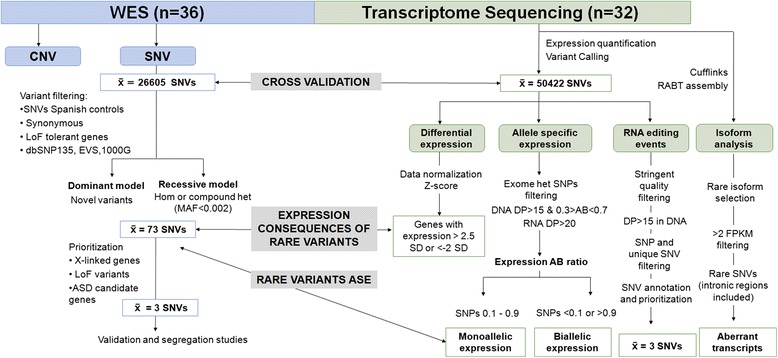
We performed whole-exome sequencing (WES) and blood cell transcriptome by RNAseq in a subset of male patients with idiopathic ASD (n = 36) in order to identify causative genes, transcriptomic alterations, and susceptibility variants.
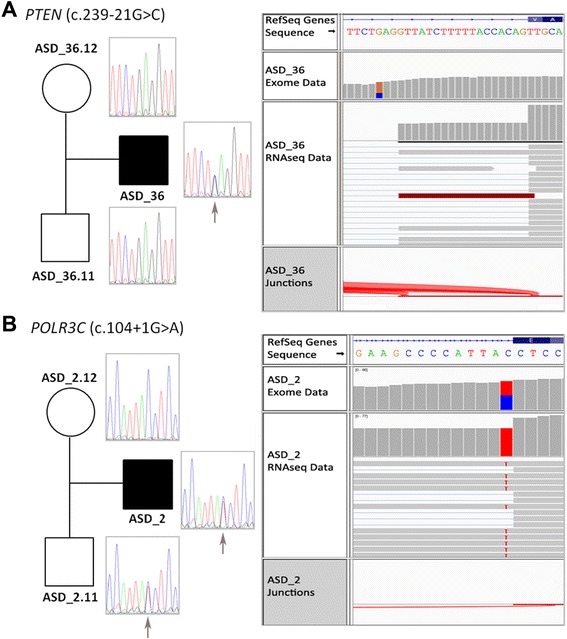
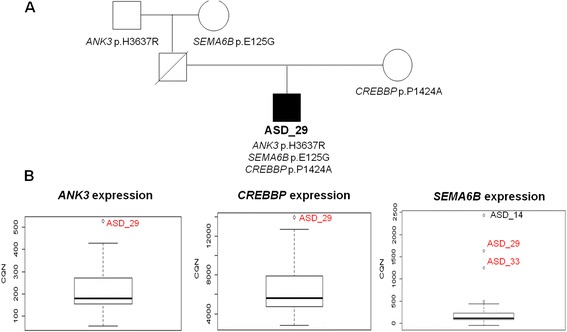
We detected likely monogenic causes in seven cases: five de novo (SCN2A, MED13L, KCNV1, CUL3, and PTEN) and two inherited X-linked variants (MAOA and CDKL5). Transcriptomic analyses allowed the identification of intronic causative mutations missed by the usual filtering of WES and revealed functional consequences of some rare mutations. These included aberrant transcripts (PTEN, POLR3C), deregulated expression in 1.7% of mutated genes (that is, SEMA6B, MECP2, ANK3, CREBBP), allele-specific expression (FUS, MTOR, TAF1C), and non-sense-mediated decay (RIT1, ALG9). The analysis of rare inherited variants showed enrichment in relevant pathways such as the PI3K-Akt signaling and the axon guidance.
Integrative analysis of WES and blood RNAseq data has proven to be an efficient strategy to identify likely monogenic forms of ASD (19% in our cohort), as well as additional rare inherited mutations that can contribute to ASD risk in a multifactorial manner. Blood transcriptomic data, besides validating 88% of expressed variants, allowed the identification of missed intronic mutations and revealed functional correlations of genetic variants, including changes in splicing, expression levels, and allelic expression.
自闭症谱系障碍(ASD)是一组具有高遗传性的神经发育障碍。最近的研究结果支持其具有高度异质性和复杂的遗传病因,包括罕见的新生突变和遗传突变、染色体重排以及双重或多重打击。
我们对一部分特发性ASD男性患者(n = 36)进行了全外显子组测序(WES)和通过RNA测序进行血细胞转录组分析,以确定致病基因、转录组改变和易感性变异。
我们在7例病例中检测到可能的单基因病因:5个新生突变(SCN2A、MED13L、KCNV1、CUL3和PTEN)和2个遗传性X连锁变异(MAOA和CDKL5)。转录组分析能够识别通常WES筛选遗漏的内含子致病突变,并揭示一些罕见突变的功能后果。这些包括异常转录本(PTEN、POLR3C)、1.7%的突变基因中表达失调(即SEMA6B、MECP2、ANK3、CREBBP)、等位基因特异性表达(FUS、MTOR、TAF1C)以及无义介导的衰变(RIT1、ALG9)。对罕见遗传变异的分析显示在相关通路中富集,如PI3K - Akt信号通路和轴突导向。
WES和血液RNA测序数据的综合分析已被证明是一种有效的策略,可识别可能的单基因形式的ASD(我们队列中的比例为19%),以及其他可能以多因素方式导致ASD风险的罕见遗传突变。血液转录组数据除了验证88%的表达变异外,还能识别遗漏的内含子突变,并揭示遗传变异的功能相关性,包括剪接、表达水平和等位基因表达的变化。