Quaiser Achim, Dufresne Alexis, Ballaud Flore, Roux Simon, Zivanovic Yvan, Colombet Jonathan, Sime-Ngando Télesphore, Francez André-Jean
UMR CNRS 6553 - ECOBIO, Université de Rennes 1 Rennes, France.
Department of Ecology and Evolutionary Biology, University of Arizona, Tucson, AZ USA.
Front Microbiol. 2015 Apr 28;6:375. doi: 10.3389/fmicb.2015.00375. eCollection 2015.
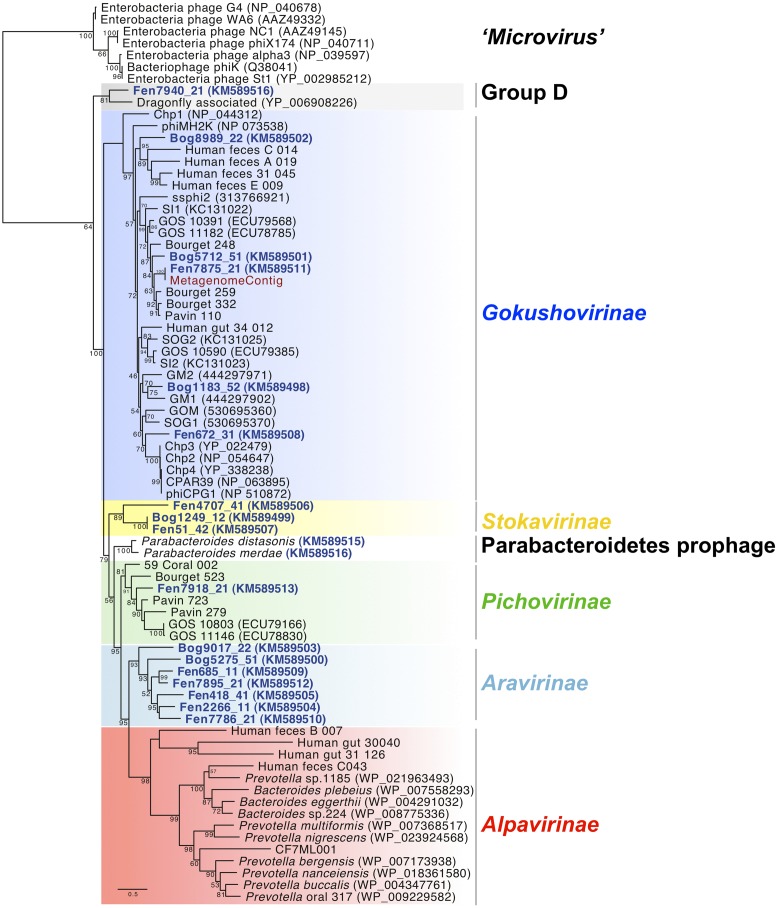
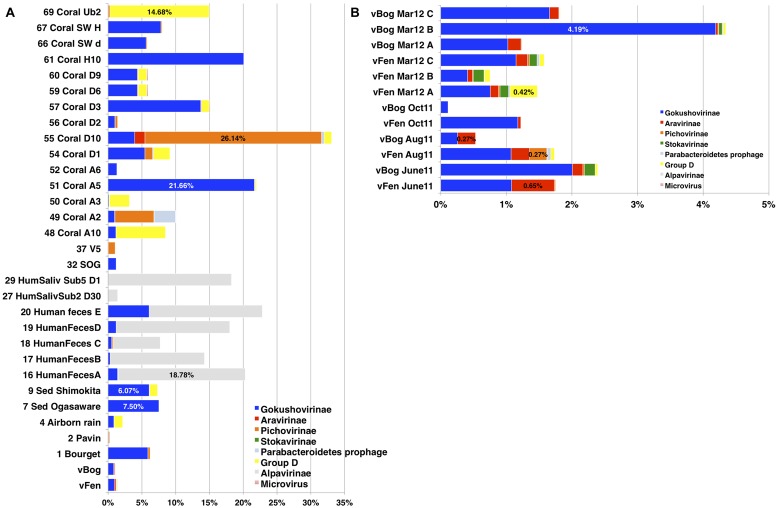
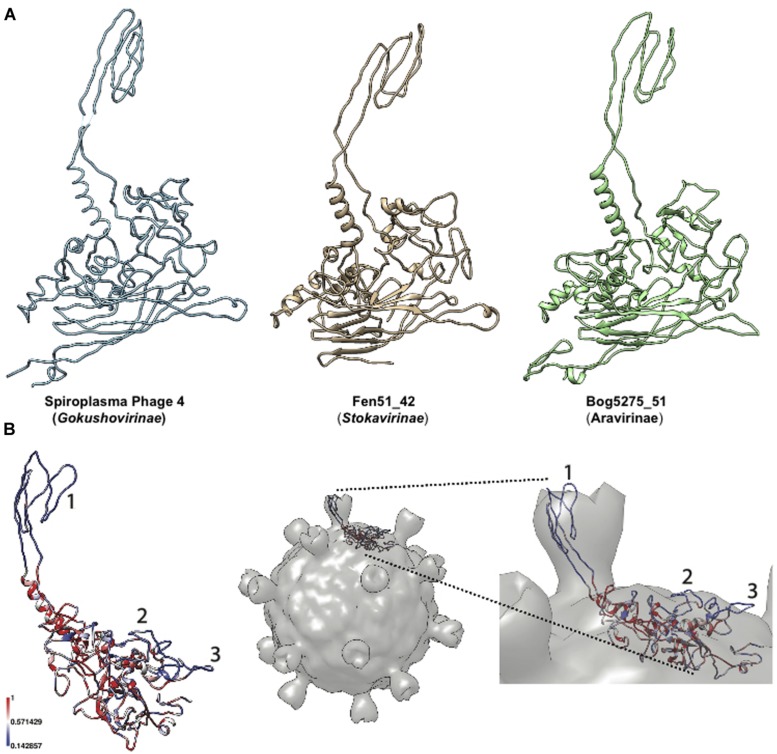
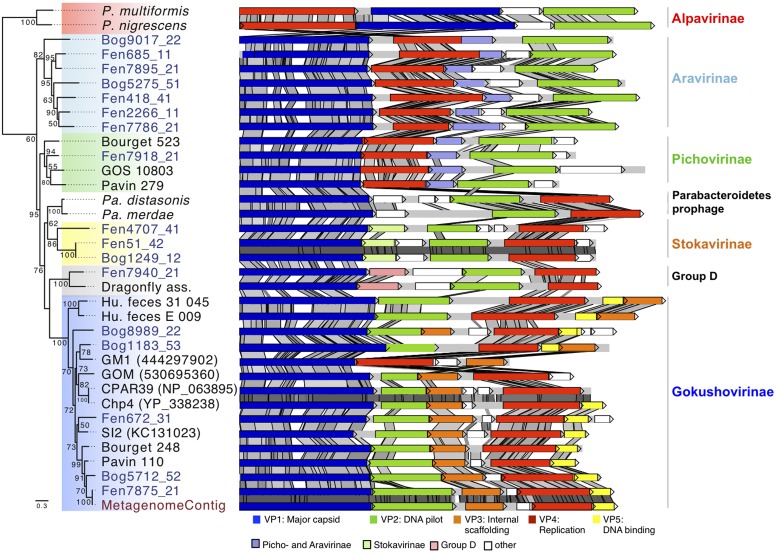
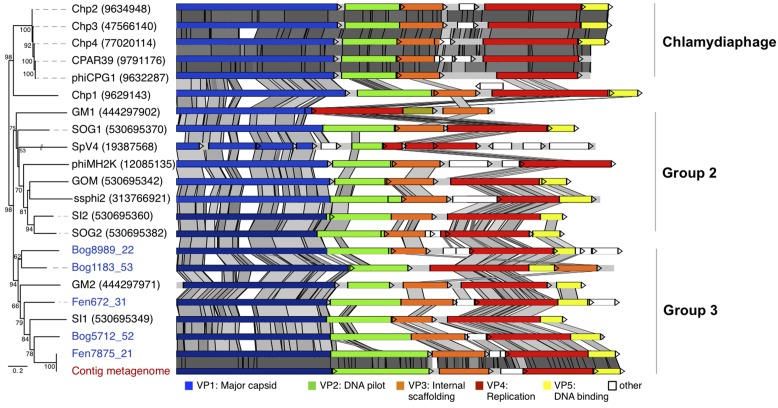
Microviridae, a family of bacteria-infecting ssDNA viruses, is one of the still poorly characterized bacteriophage groups, even though it includes phage PhiX174, one of the main models in virology for genomic and capsid structure studies. Recent studies suggest that they are diverse and well represented in marine and freshwater virioplankton as well as in human microbiomes. However, their diversity, abundance, and ecological role are completely unknown in soil ecosystems. Here we present the comparative analysis of 17 completely assembled Microviridae genomes from 12 viromes of a Sphagnum-dominated peatland. Phylogenetic analysis of the conserved major capsid protein sequences revealed the affiliation to Gokushovirinae and Pichovirinae as well as to two newly defined subfamilies, the Aravirinae and Stokavirinae. Additionally, two new distinct prophages were identified in the genomes of Parabacteroides merdae and Parabacteroides distasonis representing a potential new subfamily of Microviridae. The differentiation of the subfamilies was confirmed by gene order and similarity analysis. Relative abundance analysis using the affiliation of the major capsid protein (VP1) revealed that Gokushovirinae, followed by Aravirinae, are the most abundant Microviridae in 11 out of 12 peat viromes. Sequences matching the Gokushovirinae and Aravirinae VP1 matching sequences, respectively, accounted for up to 4.19 and 0.65% of the total number of sequences in the corresponding virome, respectively. In this study we provide new genome information of Microviridae and pave the way toward quantitative estimations of Microviridae subfamilies.
微小病毒科是一类感染细菌的单链DNA病毒,是目前仍未得到充分研究的噬菌体群体之一,尽管它包含噬菌体PhiX174,这是病毒学中用于基因组和衣壳结构研究的主要模型之一。最近的研究表明,它们具有多样性,在海洋和淡水病毒浮游生物以及人类微生物群落中都有很好的代表性。然而,它们在土壤生态系统中的多样性、丰度和生态作用却完全未知。在这里,我们对来自以泥炭藓为主的泥炭地的12个病毒宏基因组中的17个完全组装的微小病毒科基因组进行了比较分析。对保守的主要衣壳蛋白序列进行系统发育分析,揭示了它们与古 Kushovirinae 亚科、Pichovirinae 亚科以及两个新定义的亚科 Aravirinae 和 Stokavirinae 的亲缘关系。此外,在 Merdae 副拟杆菌和 Distasonis 副拟杆菌的基因组中鉴定出两个新的独特原噬菌体,它们代表了微小病毒科的一个潜在新亚科。通过基因顺序和相似性分析证实了亚科的分化。利用主要衣壳蛋白(VP1)的归属进行相对丰度分析表明,在12个泥炭病毒宏基因组中的11个中,Kushovirinae 亚科(其次是 Aravirinae 亚科)是最丰富的微小病毒科。与 Kushovirinae 亚科和 Aravirinae 亚科VP1匹配序列分别匹配的序列,分别占相应病毒宏基因组中序列总数的4.19%和0.65%。在这项研究中,我们提供了微小病毒科的新基因组信息,并为微小病毒科亚科的定量估计铺平了道路。