Pulido-Tamayo Sergio, Sánchez-Rodríguez Aminael, Swings Toon, Van den Bergh Bram, Dubey Akanksha, Steenackers Hans, Michiels Jan, Fostier Jan, Marchal Kathleen
Department of Information Technology, Ghent University, iMinds, 9050 Gent, Belgium Department of Microbial and Molecular Systems, Centre of Microbial and Plant Genetics, KU Leuven, Kasteelpark Arenberg 20, 3001 Leuven, Belgium Department of Plant Biotechnology and Bioinformatics, Ghent University, 9052 Ghent, Belgium.
Department of Microbial and Molecular Systems, Centre of Microbial and Plant Genetics, KU Leuven, Kasteelpark Arenberg 20, 3001 Leuven, Belgium Departamento de Ciencias Naturales, Universidad Técnica Particular de Loja, San Cayetano Alto S/N, EC1101608 Loja, Ecuador.
Nucleic Acids Res. 2015 Sep 18;43(16):e105. doi: 10.1093/nar/gkv478. Epub 2015 May 18.
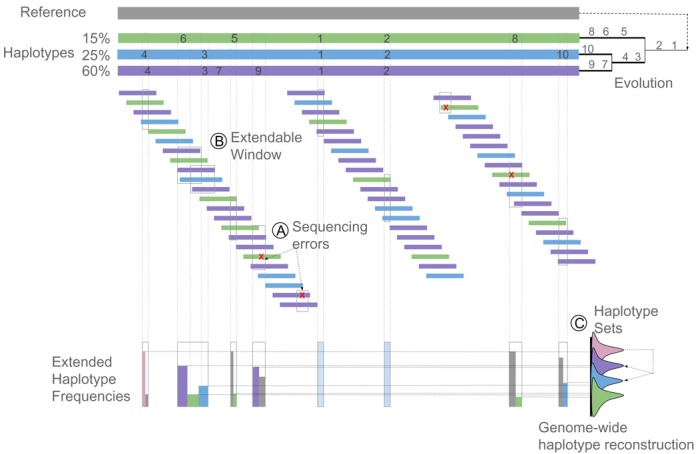
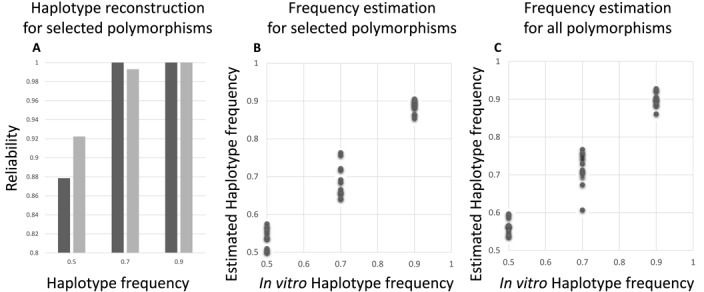
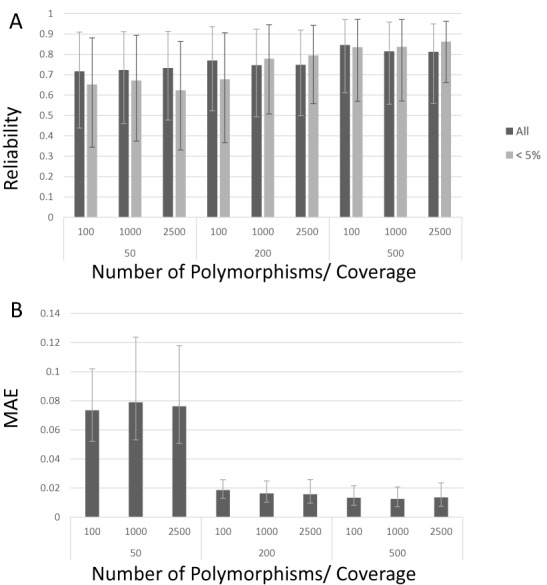
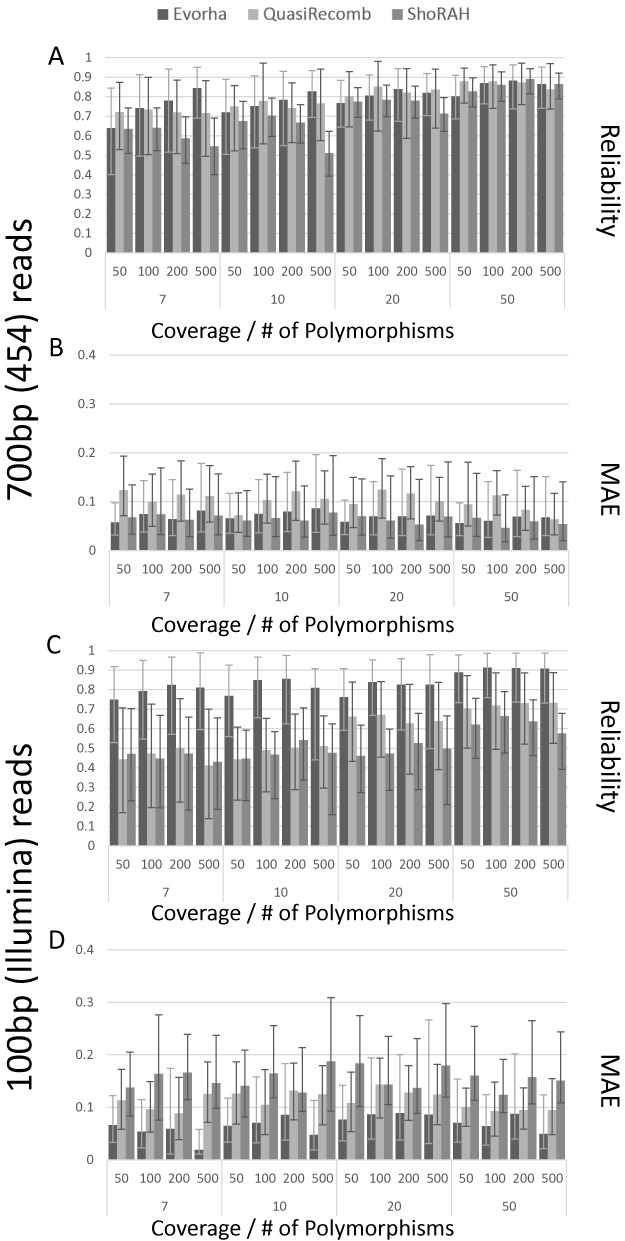
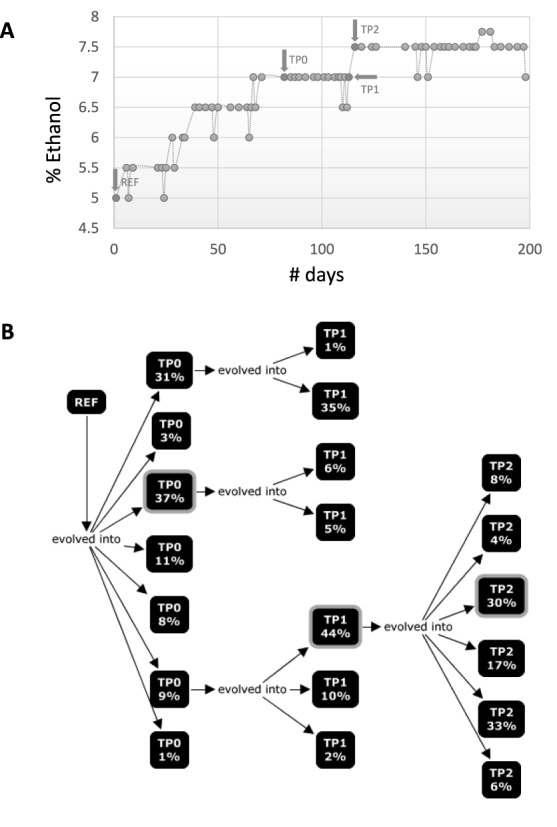
Clonal populations accumulate mutations over time, resulting in different haplotypes. Deep sequencing of such a population in principle provides information to reconstruct these haplotypes and the frequency at which the haplotypes occur. However, this reconstruction is technically not trivial, especially not in clonal systems with a relatively low mutation frequency. The low number of segregating sites in those systems adds ambiguity to the haplotype phasing and thus obviates the reconstruction of genome-wide haplotypes based on sequence overlap information.Therefore, we present EVORhA, a haplotype reconstruction method that complements phasing information in the non-empty read overlap with the frequency estimations of inferred local haplotypes. As was shown with simulated data, as soon as read lengths and/or mutation rates become restrictive for state-of-the-art methods, the use of this additional frequency information allows EVORhA to still reliably reconstruct genome-wide haplotypes. On real data, we show the applicability of the method in reconstructing the population composition of evolved bacterial populations and in decomposing mixed bacterial infections from clinical samples.
克隆群体随时间积累突变,从而产生不同的单倍型。对这样一个群体进行深度测序原则上可提供信息以重建这些单倍型及其出现频率。然而,这种重建在技术上并非易事,尤其是在突变频率相对较低的克隆系统中。这些系统中分离位点数量较少,给单倍型定相增加了不确定性,因此无法基于序列重叠信息重建全基因组单倍型。因此,我们提出了EVORhA,这是一种单倍型重建方法,它将非空读段重叠中的定相信息与推断的局部单倍型频率估计相结合。正如模拟数据所示,一旦读长和/或突变率对现有方法构成限制,使用这种额外的频率信息可使EVORhA仍能可靠地重建全基因组单倍型。在实际数据中,我们展示了该方法在重建进化细菌群体的群体组成以及分解临床样本中的混合细菌感染方面的适用性。