Pilla Kala Bharath, Leman Julia Koehler, Otting Gottfried, Huber Thomas
Research School of Chemistry, Australian National University, Canberra, ACT 2601, Australia.
Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, MD, 21218, United States of America.
PLoS One. 2015 May 18;10(5):e0127053. doi: 10.1371/journal.pone.0127053. eCollection 2015.
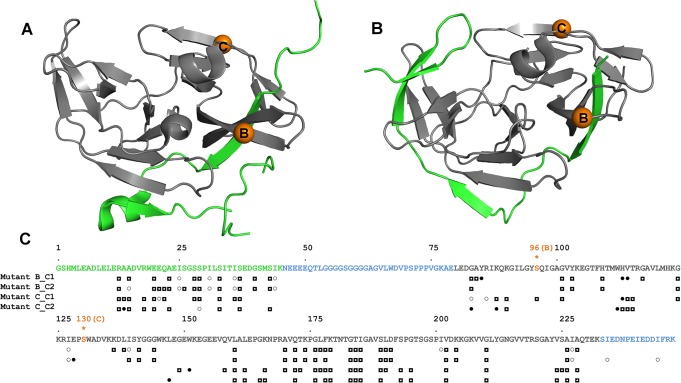
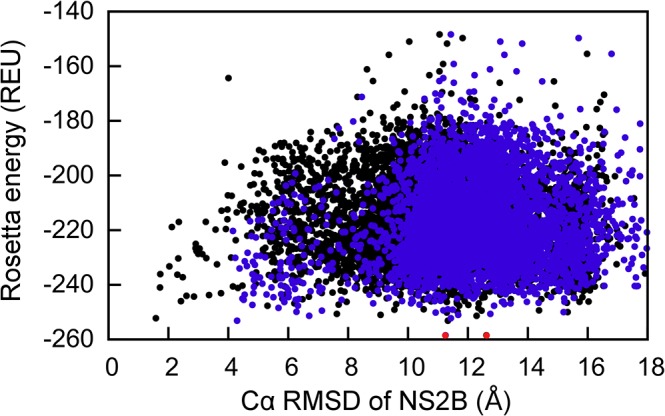
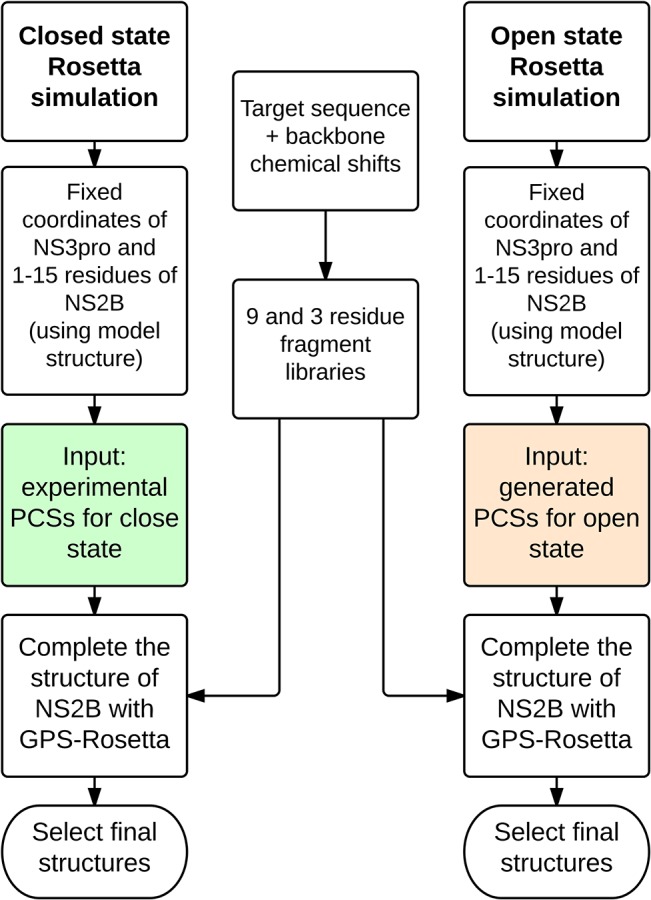
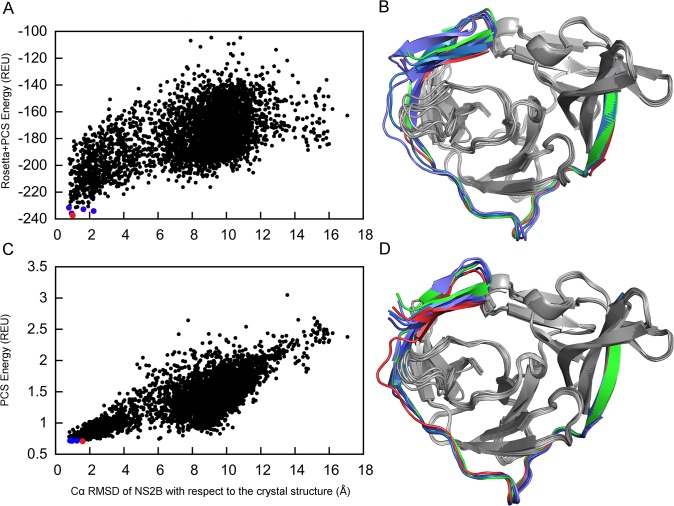
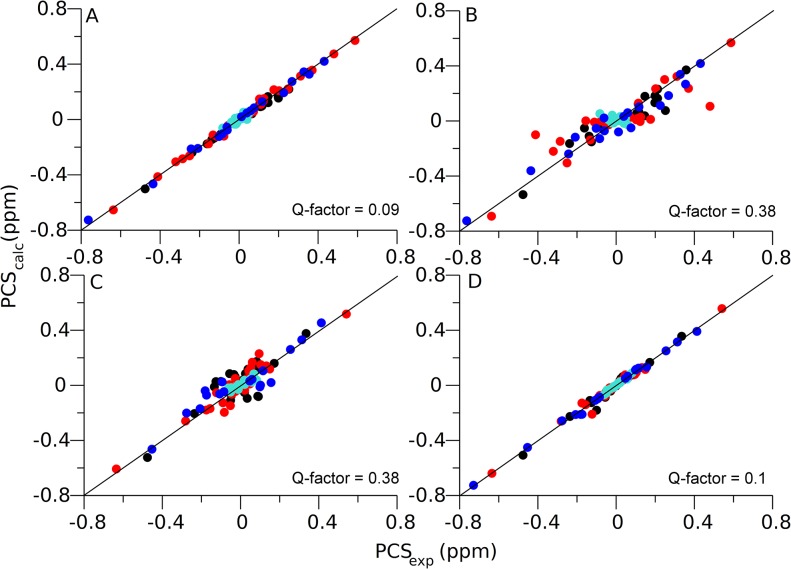
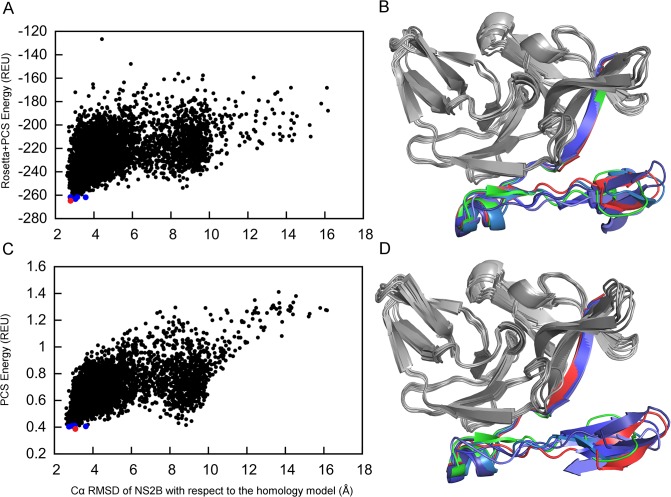
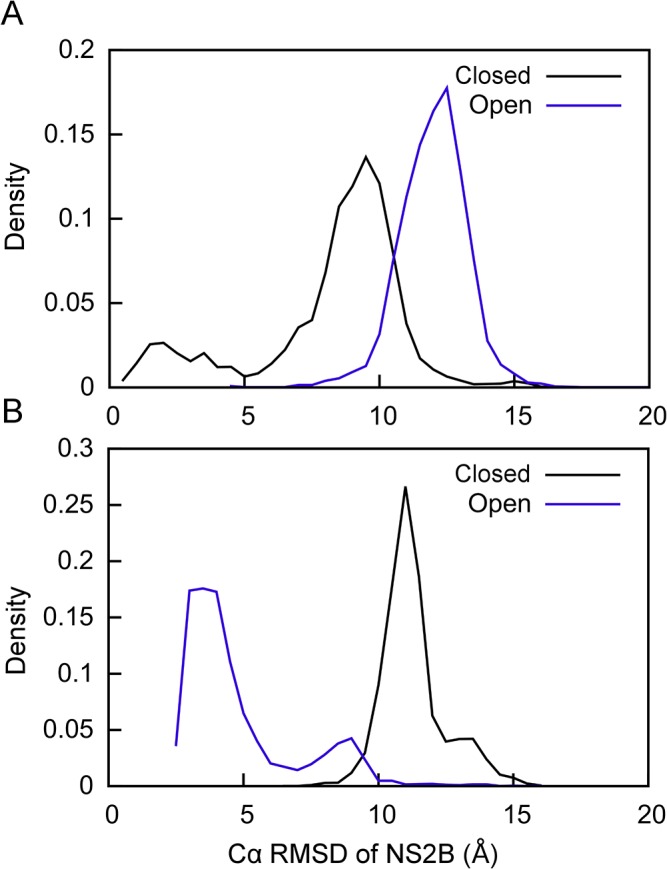
Capturing conformational changes in proteins or protein-protein complexes is a challenge for both experimentalists and computational biologists. Solution nuclear magnetic resonance (NMR) is unique in that it permits structural studies of proteins under greatly varying conditions, and thus allows us to monitor induced structural changes. Paramagnetic effects are increasingly used to study protein structures as they give ready access to rich structural information of orientation and long-range distance restraints from the NMR signals of backbone amides, and reliable methods have become available to tag proteins with paramagnetic metal ions site-specifically and at multiple sites. In this study, we show how sparse pseudocontact shift (PCS) data can be used to computationally model conformational states in a protein system, by first identifying core structural elements that are not affected by the environmental change, and then computationally completing the remaining structure based on experimental restraints from PCS. The approach is demonstrated on a 27 kDa two-domain NS2B-NS3 protease system of the dengue virus serotype 2, for which distinct closed and open conformational states have been observed in crystal structures. By changing the input PCS data, the observed conformational states in the dengue virus protease are reproduced without modifying the computational procedure. This data driven Rosetta protocol enables identification of conformational states of a protein system, which are otherwise difficult to obtain either experimentally or computationally.
捕捉蛋白质或蛋白质-蛋白质复合物中的构象变化,对实验人员和计算生物学家来说都是一项挑战。溶液核磁共振(NMR)的独特之处在于,它允许在各种不同条件下对蛋白质进行结构研究,从而使我们能够监测诱导的结构变化。顺磁效应越来越多地用于研究蛋白质结构,因为它们能从主链酰胺的NMR信号中轻易获取丰富的取向和长程距离限制的结构信息,并且已经有了可靠的方法来将顺磁金属离子位点特异性地标记在蛋白质的多个位点上。在本研究中,我们展示了如何通过首先识别不受环境变化影响的核心结构元件,然后根据来自伪接触位移(PCS)的实验限制进行计算来完成其余结构,从而利用稀疏的伪接触位移(PCS)数据对蛋白质系统中的构象状态进行计算建模。该方法在登革热病毒2型的27 kDa双结构域NS2B-NS3蛋白酶系统上得到了验证,在该系统的晶体结构中观察到了不同的封闭和开放构象状态。通过改变输入的PCS数据,在不修改计算程序的情况下再现了登革热病毒蛋白酶中观察到的构象状态。这种数据驱动的Rosetta协议能够识别蛋白质系统的构象状态,而这些构象状态通过实验或计算方法很难获得。