Hartman Joshua D, Neubauer Thomas J, Caulkins Bethany G, Mueller Leonard J, Beran Gregory J O
Department of Chemistry, University of California at Riverside, Riverside, CA, 92521, USA.
J Biomol NMR. 2015 Jul;62(3):327-40. doi: 10.1007/s10858-015-9947-2. Epub 2015 May 21.
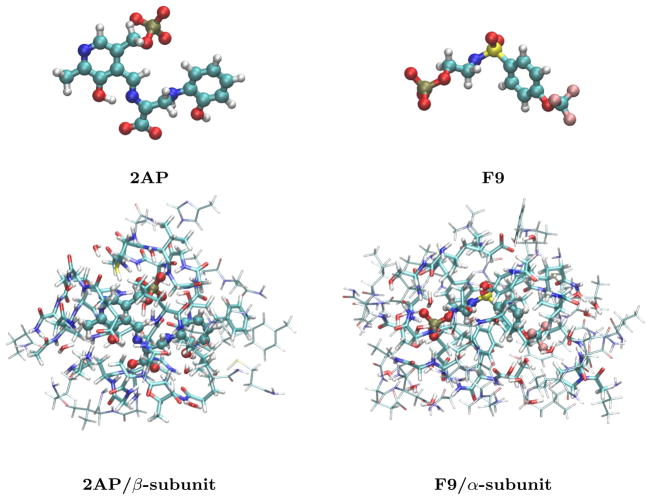
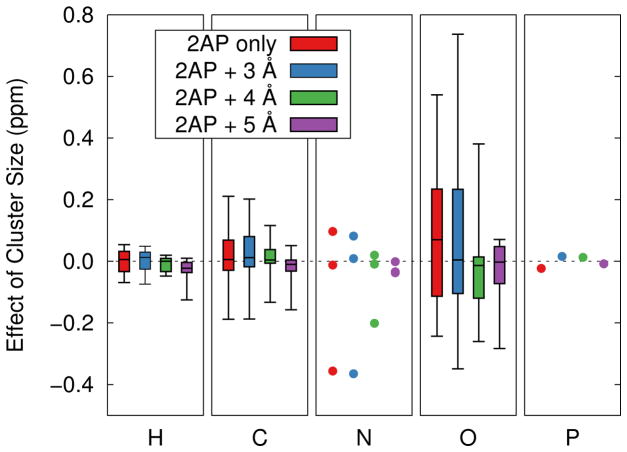
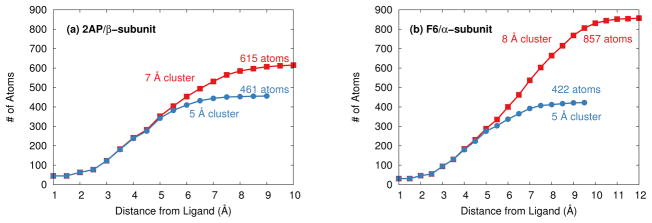
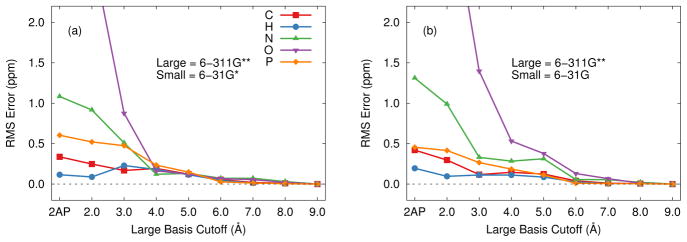
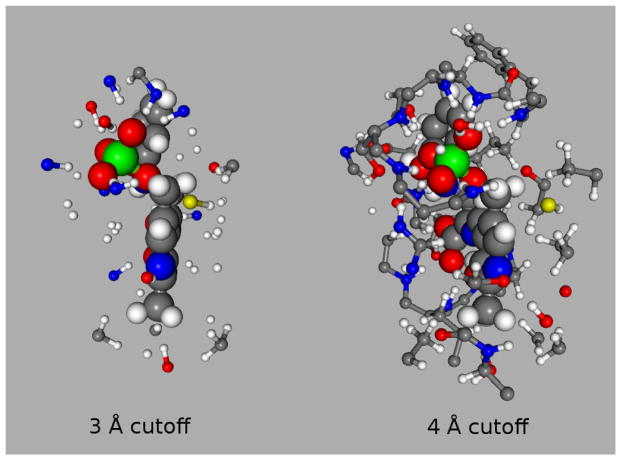
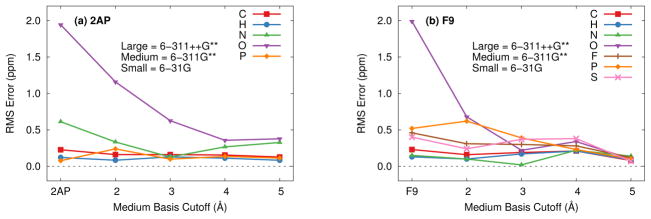
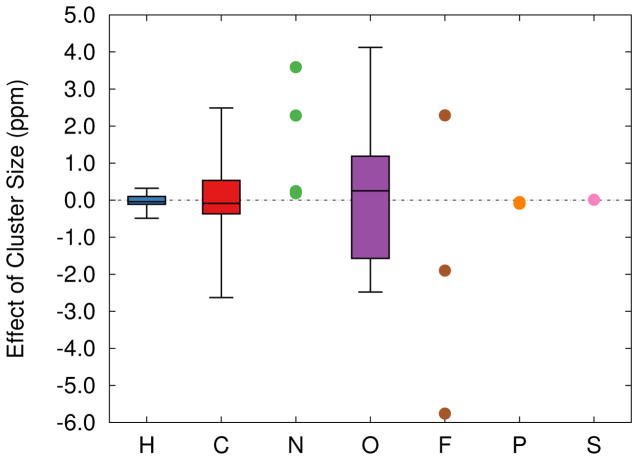
Ab initio chemical shielding calculations greatly facilitate the interpretation of nuclear magnetic resonance (NMR) chemical shifts in biological systems, but the large sizes of these systems requires approximations in the chemical models used to represent them. Achieving good convergence in the predicted chemical shieldings is necessary before one can unravel how other complex structural and dynamical factors affect the NMR measurements. Here, we investigate how to balance trade-offs between using a better basis set or a larger cluster model for predicting the chemical shieldings of the substrates in two representative examples of protein-substrate systems involving different domains in tryptophan synthase: the N-(4'-trifluoromethoxybenzoyl)-2-aminoethyl phosphate (F9) ligand which binds in the α active site, and the 2-aminophenol quinonoid intermediate formed in the β active site. We first demonstrate that a chemically intuitive three-layer, locally dense basis model that uses a large basis on the substrate, a medium triple-zeta basis to describe its hydrogen-bonding partners and/or surrounding van der Waals cavity, and a crude basis set for more distant atoms provides chemical shieldings in good agreement with much more expensive large basis calculations. Second, long-range quantum mechanical interactions are important, and one can accurately estimate them as a small-basis correction to larger-basis calculations on a smaller cluster. The combination of these approaches enables one to perform density functional theory NMR chemical shift calculations in protein systems that are well-converged with respect to both basis set and cluster size.
从头算化学屏蔽计算极大地促进了对生物系统中核磁共振(NMR)化学位移的解释,但这些系统的大尺寸要求在用于表示它们的化学模型中采用近似方法。在揭示其他复杂的结构和动力学因素如何影响NMR测量之前,在预测的化学屏蔽中实现良好的收敛是必要的。在这里,我们研究了在两个涉及色氨酸合酶不同结构域的蛋白质-底物系统的代表性例子中,如何在使用更好的基组或更大的簇模型来预测底物的化学屏蔽之间进行权衡:结合在α活性位点的N-(4'-三氟甲氧基苯甲酰基)-2-氨基乙基磷酸酯(F9)配体,以及在β活性位点形成的2-氨基酚醌中间体。我们首先证明,一种具有化学直观性的三层局部密集基模型,即在底物上使用大基组,用中等三重ζ基组来描述其氢键伙伴和/或周围的范德华腔,并用粗糙基组来描述更远的原子,所提供的化学屏蔽与更昂贵的大基组计算结果吻合良好。其次,长程量子力学相互作用很重要,并且可以将其作为对较小簇上更大基组计算的小基组校正来准确估计。这些方法的结合使人们能够在蛋白质系统中进行密度泛函理论NMR化学位移计算,该计算在基组和簇大小方面都具有良好的收敛性。