Birk Alexander V, Chao Wesley M, Liu Shaoyi, Soong Yi, Szeto Hazel H
Department of Pharmacology, Joan and Sanford I. Weill Medical College of Cornell University, New York, NY 10065, USA; Research Program in Mitochondrial Therapeutics, Joan and Sanford I. Weill Medical College of Cornell University, New York, NY 10065, USA.
Department of Pharmacology, Joan and Sanford I. Weill Medical College of Cornell University, New York, NY 10065, USA; Research Program in Mitochondrial Therapeutics, Joan and Sanford I. Weill Medical College of Cornell University, New York, NY 10065, USA.
Biochim Biophys Acta. 2015 Oct;1847(10):1075-84. doi: 10.1016/j.bbabio.2015.06.006. Epub 2015 Jun 10.
It was recently suggested that electron flow into cyt c, coupled with ROS generation, oxidizes cyt c Met(80) to Met(80) sulfoxide (Met-O) in isolated hearts after ischemia-reperfusion, and converts cyt c to a peroxidase. We hypothesize that ischemia disrupts Met(80)-Fe ligation of cyt c, forming pentacoordinated heme Fe(2+), which inhibits electron transport (ET) and promotes oxygenase activity.
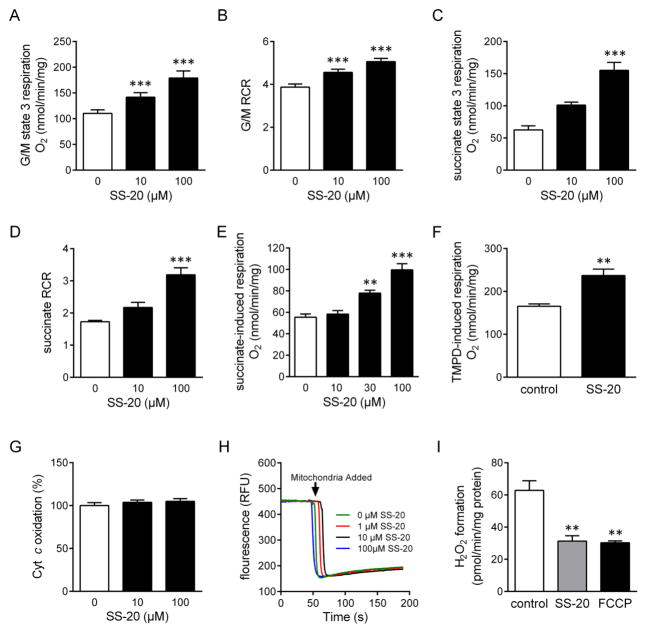
SS-20 (Phe-D-Arg-Phe-Lys-NH2) was used to demonstrate the role of Met(80)-Fe ligation in ischemia. Mitochondria were isolated from ischemic rat kidneys to determine sites of respiratory inhibition. Mitochondrial cyt c and cyt c Met-O were quantified by western blot, and cristae architecture was examined by electron microscopy.
Biochemical and structural studies showed that SS-20 selectively targets cardiolipin (CL) and protects Met(80)-Fe ligation in cyt c. Ischemic mitochondria showed 17-fold increase in Met-O cyt c, and dramatic cristaeolysis. Loss of cyt c was associated with proteolytic degradation of OPA1. Ischemia significantly inhibited ET initiated by direct reduction of cyt c and coupled respiration. All changes were prevented by SS-20.
Our results show that ischemia disrupts the Met(80)-Fe ligation of cyt c resulting in the formation of a globin-like pentacoordinated heme Fe(2+) that inhibits ET, and converts cyt c into an oxygenase to cause CL peroxidation and proteolytic degradation of OPA1, resulting in cyt c release.
Cyt c heme structure represents a novel target for minimizing ischemic injury. SS-20, which we show to selectively target CL and protect the Met(80)-Fe ligation, minimizes ischemic injury and promotes ATP recovery.
最近有研究表明,在缺血再灌注后的离体心脏中,电子流入细胞色素c并伴随活性氧生成,会将细胞色素c的蛋氨酸(Met)80氧化为甲硫氨酸亚砜(Met - O),并将细胞色素c转化为过氧化物酶。我们推测,缺血会破坏细胞色素c的Met(80)-铁配体,形成五配位血红素铁(Ⅱ),从而抑制电子传递(ET)并促进加氧酶活性。
使用SS - 20(苯丙氨酸 - D - 精氨酸 - 苯丙氨酸 - 赖氨酸 - NH₂)来证明Met(80)-铁配体在缺血中的作用。从缺血大鼠肾脏中分离出线粒体,以确定呼吸抑制位点。通过蛋白质印迹法对线粒体细胞色素c和细胞色素c Met - O进行定量,并通过电子显微镜检查嵴结构。
生化和结构研究表明,SS - 20选择性靶向心磷脂(CL)并保护细胞色素c中的Met(80)-铁配体。缺血线粒体中细胞色素c Met - O增加了17倍,并出现明显的嵴溶解。细胞色素c的丢失与OPA1的蛋白水解降解有关。缺血显著抑制了由细胞色素c直接还原引发的电子传递和偶联呼吸。所有这些变化都被SS - 20阻止。
我们的结果表明,缺血会破坏细胞色素c的Met(80)-铁配体,导致形成类似球蛋白的五配位血红素铁(Ⅱ),从而抑制电子传递,并将细胞色素c转化为加氧酶,导致心磷脂过氧化和OPA1的蛋白水解降解,从而导致细胞色素c释放。
细胞色素c血红素结构是将缺血性损伤降至最低的新靶点。我们发现SS - 20选择性靶向心磷脂并保护Met(80)-铁配体,可将缺血性损伤降至最低并促进ATP恢复。