Akinrinade Oyediran, Ollila Laura, Vattulainen Sanna, Tallila Jonna, Gentile Massimiliano, Salmenperä Pertteli, Koillinen Hannele, Kaartinen Maija, Nieminen Markku S, Myllykangas Samuel, Alastalo Tero-Pekka, Koskenvuo Juha W, Heliö Tiina
Children's Hospital, Institute of Clinical Medicine, Helsinki University Central Hospital, University of Helsinki, Helsinki, Finland Institute of Biomedicine, University of Helsinki, Helsinki, Finland.
Heart and Lung Center HUCH, University of Helsinki, Helsinki, Finland.
Eur Heart J. 2015 Sep 7;36(34):2327-37. doi: 10.1093/eurheartj/ehv253. Epub 2015 Jun 17.
Despite our increased understanding of the genetic basis of dilated cardiomyopathy (DCM), the clinical utility and yield of clinically meaningful findings of comprehensive next-generation sequencing (NGS)-based genetic diagnostics in DCM has been poorly described. We utilized a high-quality oligonucleotide-selective sequencing (OS-Seq)-based targeted sequencing panel to investigate the genetic landscape of DCM in Finnish population and to evaluate the utility of OS-Seq technology as a novel comprehensive diagnostic tool.
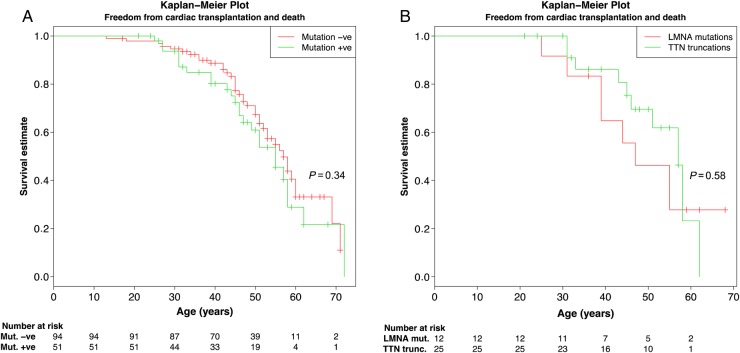
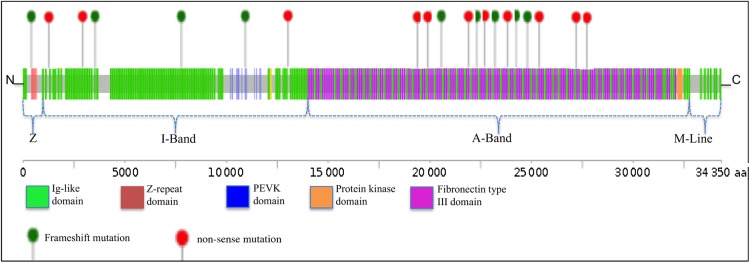
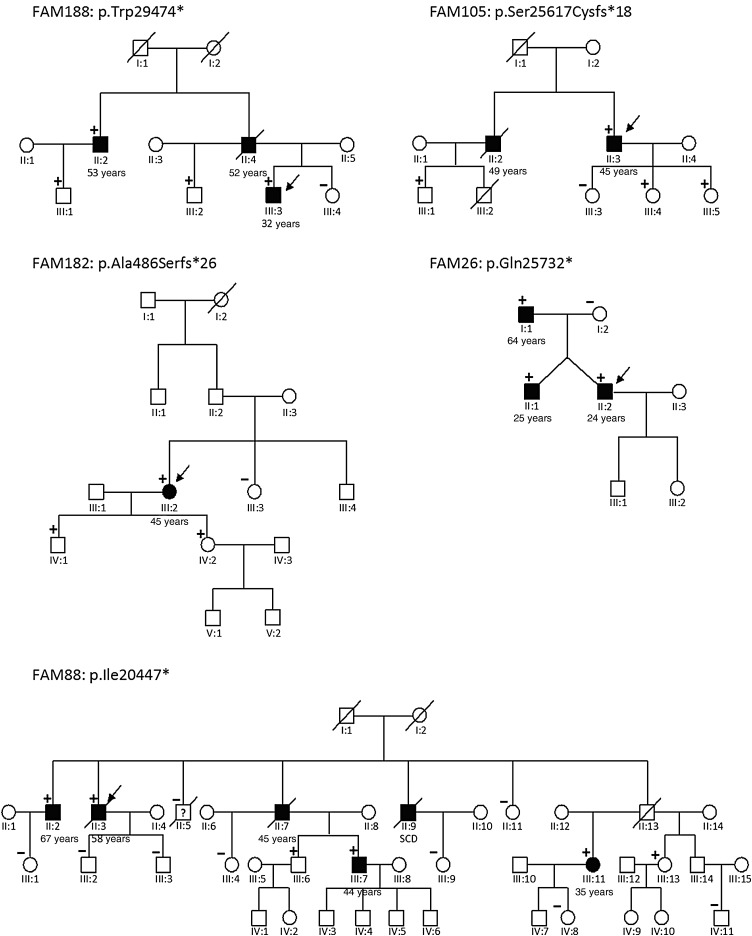
Using OS-Seq, we targeted and sequenced the coding regions and splice junctions of 101 genes associated with cardiomyopathies in 145 unrelated Finnish patients with DCM. We developed effective bioinformatic variant filtering strategy and implemented strict variant classification scheme to reveal diagnostic yield and genotype-phenotype correlations. Implemented OS-Seq technology provided high coverage of the target region (median coverage 410× and 99.42% of the nucleotides were sequenced at least 15× read depth). Diagnostic yield was 35.2% (familial 47.6% and sporadic 25.6%, P = 0.004) when both pathogenic and likely pathogenic variants are considered as disease causing. Of these, 20 (53%) were titin (TTN) truncations (non-sense and frameshift) affecting all TTN transcripts. TTN truncations accounted for 20.6% and 14.6% of the familial and sporadic DCM cases, respectively.
Panel-based, high-quality NGS enables high diagnostic yield especially in the familial form of DCM, and bioinformatic variant filtering is a reliable step in the process of interpretation of genomic data in a clinical setting.
尽管我们对扩张型心肌病(DCM)的遗传基础有了更多了解,但基于新一代测序(NGS)的全面基因诊断在DCM中具有临床意义的发现的临床实用性和产出情况却鲜有描述。我们利用基于高质量寡核苷酸选择性测序(OS-Seq)的靶向测序面板来研究芬兰人群中DCM的遗传图谱,并评估OS-Seq技术作为一种新型全面诊断工具的实用性。
我们使用OS-Seq技术,对145名无亲缘关系的芬兰DCM患者中与心肌病相关的101个基因的编码区和剪接位点进行靶向测序。我们开发了有效的生物信息学变异过滤策略,并实施了严格的变异分类方案,以揭示诊断产出和基因型-表型相关性。所采用的OS-Seq技术对目标区域具有高覆盖率(中位覆盖率410×,99.42%的核苷酸测序深度至少为15×)。当将致病和可能致病的变异都视为致病因素时,诊断产出为35.2%(家族性为47.6%,散发性为25.6%,P = 0.004)。其中,20个(53%)是肌联蛋白(TTN)截短变异(无义突变和移码突变),影响所有TTN转录本。TTN截短变异分别占家族性和散发性DCM病例的20.6%和14.6%。
基于面板的高质量NGS能够实现高诊断产出,尤其是在家族性DCM中,并且生物信息学变异过滤是临床环境中基因组数据解读过程中的可靠步骤。