Jiang Yi-Zhou, Manduchi Elisabetta, Stoeckert Christian J, Davies Peter F
Department of Pathology & Laboratory Medicine and Institute for Medicine & Engineering, Perelman School of Medicine, University of Pennsylvania, 1010 Vagelos Building, 3340 Smith Walk, Philadelphia, PA, 19104, USA.
Department of Genetics and Institute for Biomedical Informatics, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, 19104, USA.
BMC Genomics. 2015 Jul 7;16:506. doi: 10.1186/s12864-015-1656-4.
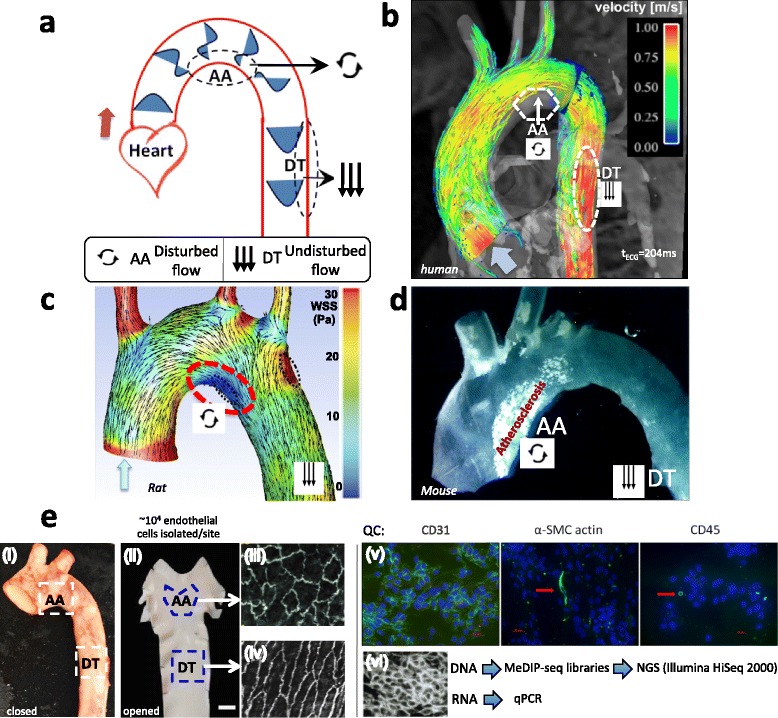
Atherosclerosis is a heterogeneously distributed disease of arteries in which the endothelium plays an important central role. Spatial transcriptome profiling of endothelium in pre-lesional arteries has demonstrated differential phenotypes primed for athero-susceptibility at hemodynamic sites associated with disturbed blood flow. DNA methylation is a powerful epigenetic regulator of endothelial transcription recently associated with flow characteristics. We investigated differential DNA methylation in flow region-specific aortic endothelial cells in vivo in adult domestic male and female swine.
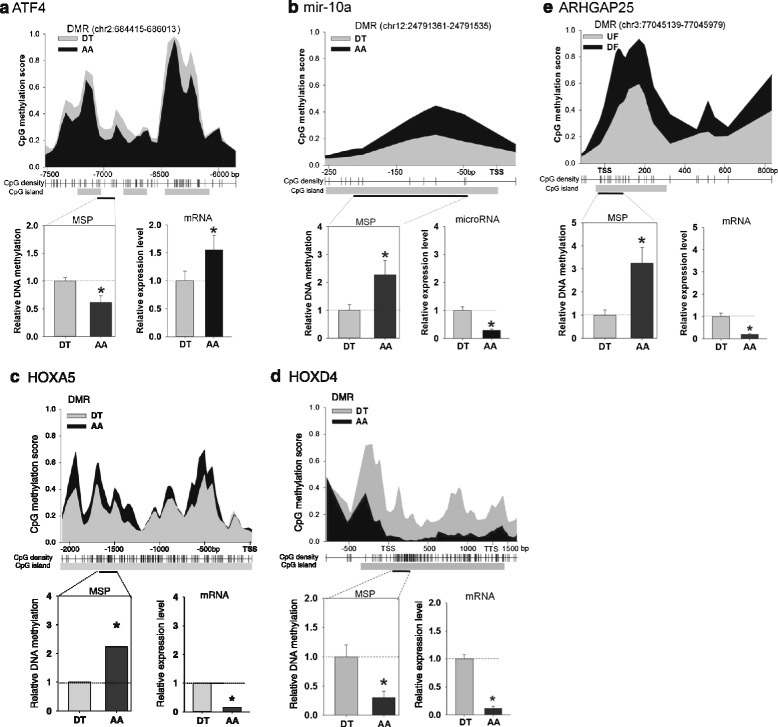
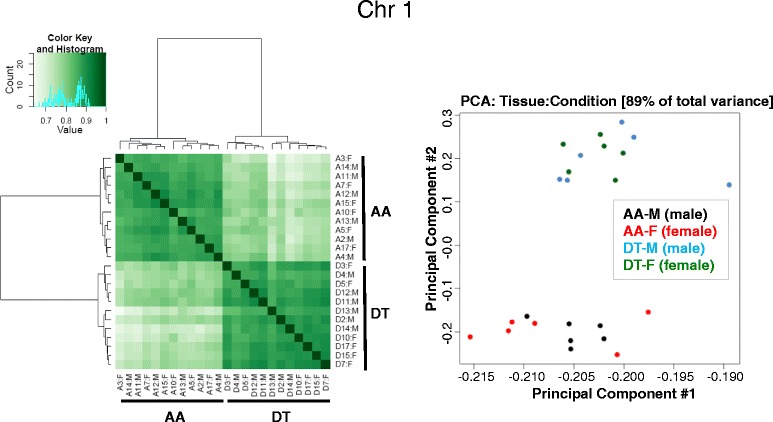
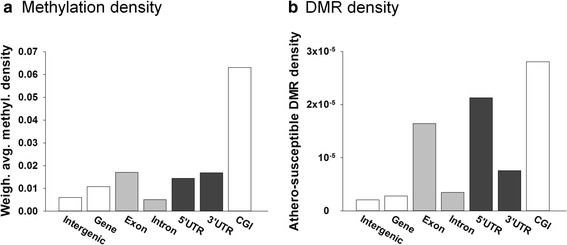
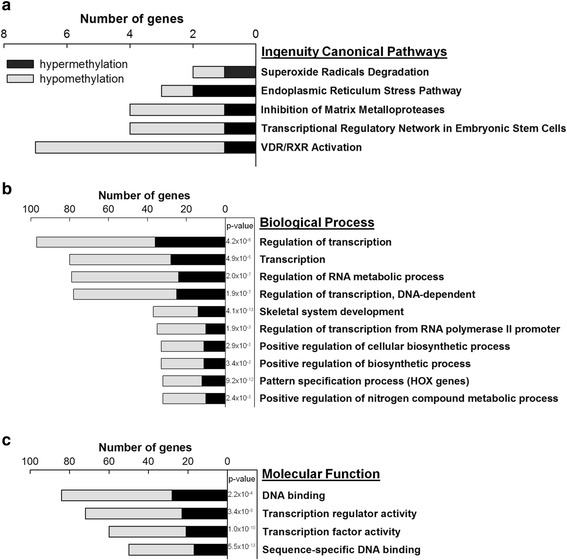
Genome-wide DNA methylation was profiled in endothelial cells (EC) isolated from two robust locations of differing patho-susceptibility:--an athero-susceptible site located at the inner curvature of the aortic arch (AA) and an athero-protected region in the descending thoracic (DT) aorta. Complete methylated DNA immunoprecipitation sequencing (MeDIP-seq) identified over 5500 endothelial differentially methylated regions (DMRs). DMR density was significantly enriched in exons and 5'UTR sequences of annotated genes, 60 of which are linked to cardiovascular disease. The set of DMR-associated genes was enriched in transcriptional regulation, pattern specification HOX loci, oxidative stress and the ER stress adaptive pathway, all categories linked to athero-susceptible endothelium. Examination of the relationship between DMR and mRNA in HOXA genes demonstrated a significant inverse relationship between CpG island promoter methylation and gene expression. Methylation-specific PCR (MSP) confirmed differential CpG methylation of HOXA genes, the ER stress gene ATF4, inflammatory regulator microRNA-10a and ARHGAP25 that encodes a negative regulator of Rho GTPases involved in cytoskeleton remodeling. Gender-specific DMRs associated with ciliogenesis that may be linked to defects in cilia development were also identified in AA DMRs.
An endothelial methylome analysis identifies epigenetic DMR characteristics associated with transcriptional regulation in regions of atherosusceptibility in swine aorta in vivo. The data represent the first methylome blueprint for spatio-temporal analyses of lesion susceptibility predisposing to endothelial dysfunction in complex flow environments in vivo.
动脉粥样硬化是一种动脉分布不均一的疾病,其中内皮发挥着重要的核心作用。对病变前动脉内皮进行空间转录组分析,已证明在与血流紊乱相关的血流动力学部位,内皮细胞存在易患动脉粥样硬化的不同表型。DNA甲基化是一种强大的内皮转录表观遗传调节因子,最近发现其与血流特征有关。我们研究了成年家猪体内血流区域特异性主动脉内皮细胞中的差异DNA甲基化。
对从两个病理易感性不同的部位分离出的内皮细胞(EC)进行全基因组DNA甲基化分析:一个是位于主动脉弓内曲率处的动脉粥样硬化易感部位(AA),另一个是胸降主动脉中的动脉粥样硬化保护区域(DT)。全基因组甲基化DNA免疫沉淀测序(MeDIP-seq)鉴定出超过5500个内皮细胞差异甲基化区域(DMR)。DMR密度在外显子和注释基因的5'UTR序列中显著富集,其中60个与心血管疾病相关。与DMR相关的基因集在转录调控、模式规范HOX基因座、氧化应激和内质网应激适应途径中富集,所有这些类别都与易患动脉粥样硬化的内皮细胞有关。对HOXA基因中DMR与mRNA之间关系的研究表明,CpG岛启动子甲基化与基因表达之间存在显著的负相关。甲基化特异性PCR(MSP)证实了HOXA基因、内质网应激基因ATF4、炎症调节因子微小RNA-10a和ARHGAP25(编码参与细胞骨架重塑的Rho GTPases负调节因子)的差异CpG甲基化。在AA DMR中还鉴定出了与纤毛发生相关的性别特异性DMR,这些DMR可能与纤毛发育缺陷有关。
内皮甲基化组分析确定了与体内猪主动脉动脉粥样硬化易感区域转录调控相关的表观遗传DMR特征。这些数据代表了首个甲基化组蓝图,用于在体内复杂血流环境中对易导致内皮功能障碍的病变易感性进行时空分析。