Muller Etienne, Brault Baptiste, Holmes Allyson, Legros Angelina, Jeannot Emmanuelle, Campitelli Maura, Rousselin Antoine, Goardon Nicolas, Frébourg Thierry, Krieger Sophie, Crouet Hubert, Nicolas Alain, Sastre Xavier, Vaur Dominique, Castéra Laurent
Department of Cancer Biology and Genetics, CCC François Baclesse, Caen, France.
Inserm U1079, Rouen, France.
Cancer Med. 2015 Oct;4(10):1484-93. doi: 10.1002/cam4.492. Epub 2015 Jul 8.
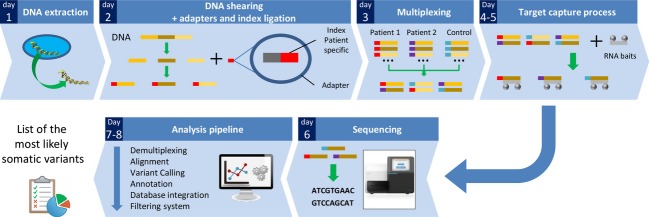
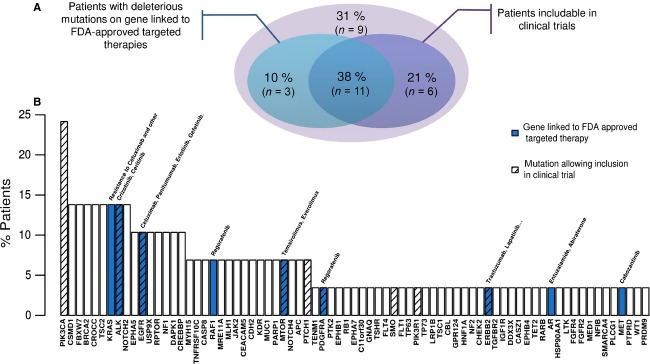
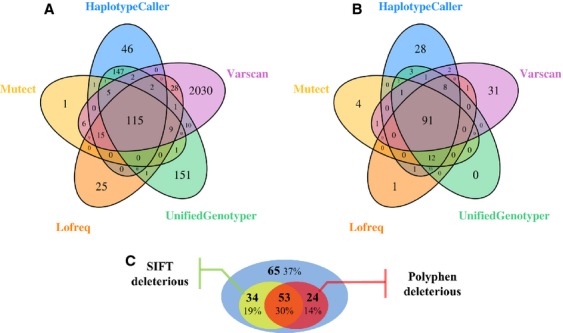
Cancer treatment is facing major evolution since the advent of targeted therapies. Building genetic profiles could predict sensitivity or resistance to these therapies and highlight disease-specific abnormalities, supporting personalized patient care. In the context of biomedical research and clinical diagnosis, our laboratory has developed an oncogenic panel comprised of 226 genes and a dedicated bioinformatic pipeline to explore somatic mutations in cervical carcinomas, using high-throughput sequencing. Twenty-nine tumors were sequenced for exons within 226 genes. The automated pipeline used includes a database and a filtration system dedicated to identifying mutations of interest and excluding false positive and germline mutations. One-hundred and seventy-six total mutational events were found among the 29 tumors. Our cervical tumor mutational landscape shows that most mutations are found in PIK3CA (E545K, E542K) and KRAS (G12D, G13D) and others in FBXW7 (R465C, R505G, R479Q). Mutations have also been found in ALK (V1149L, A1266T) and EGFR (T259M). These results showed that 48% of patients display at least one deleterious mutation in genes that have been already targeted by the Food and Drug Administration approved therapies. Considering deleterious mutations, 59% of patients could be eligible for clinical trials. Sequencing hundreds of genes in a clinical context has become feasible, in terms of time and cost. In the near future, such an analysis could be a part of a battery of examinations along the diagnosis and treatment of cancer, helping to detect sensitivity or resistance to targeted therapies and allow advancements towards personalized oncology.
自靶向治疗出现以来,癌症治疗正面临重大变革。构建基因图谱可以预测对这些治疗的敏感性或耐药性,并突出疾病特异性异常,从而支持个性化的患者护理。在生物医学研究和临床诊断的背景下,我们的实验室开发了一个由226个基因组成的致癌基因panel以及一个专门的生物信息学流程,用于通过高通量测序探索宫颈癌中的体细胞突变。对29个肿瘤的226个基因的外显子进行了测序。所使用的自动化流程包括一个数据库和一个过滤系统,专门用于识别感兴趣的突变并排除假阳性和种系突变。在这29个肿瘤中总共发现了176个突变事件。我们的宫颈肿瘤突变图谱显示,大多数突变发生在PIK3CA(E545K、E542K)和KRAS(G12D、G13D)中,其他的发生在FBXW7(R465C、R505G、R479Q)中。在ALK(V1149L、A1266T)和EGFR(T259M)中也发现了突变。这些结果表明,48%的患者在已被美国食品药品监督管理局批准的治疗所靶向的基因中至少有一个有害突变。考虑到有害突变,59%的患者可能符合临床试验条件。在临床环境中对数百个基因进行测序在时间和成本方面已变得可行。在不久的将来,这样的分析可能会成为癌症诊断和治疗一系列检查的一部分,有助于检测对靶向治疗的敏感性或耐药性,并推动个性化肿瘤学的发展。