Lim Jing-Quan, Tennakoon Chandana, Guan Peiyong, Sung Wing-Kin
Department of Computer Science, National University of Singapore, Singapore 117417 Laboratory of Cancer Epigenome, Division of Medical Sciences, National Cancer Centre Singapore, Singapore 169610.
Department of Computer Science, National University of Singapore, Singapore 117417 NUS Graduate School for Integrative Sciences and Engineering, (CeLS), #05-01, 28 Medical Drive, Singapore 117456 Department of Computational and Systems Biology, Genome Institute of Singapore, Singapore 138672 UAE University, PO Box 17551, Al Ain, UAE.
Nucleic Acids Res. 2015 Sep 18;43(16):e107. doi: 10.1093/nar/gkv533. Epub 2015 Jul 13.
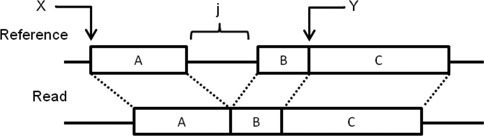
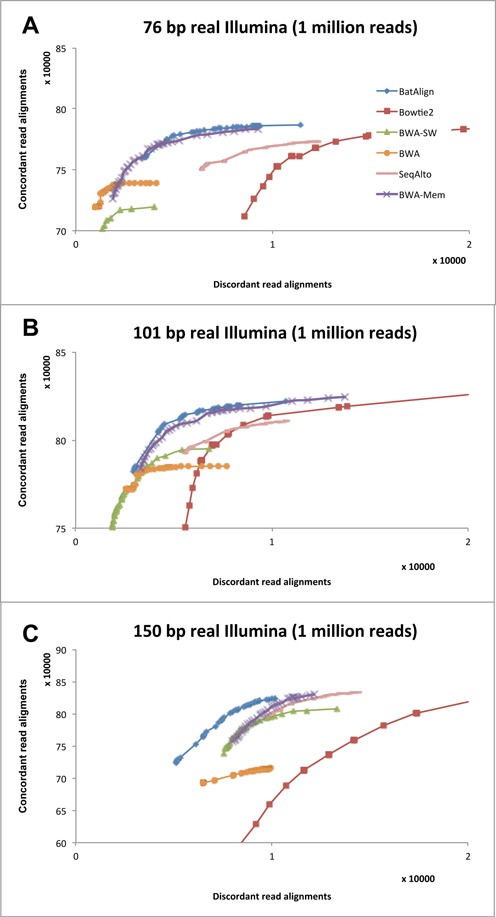
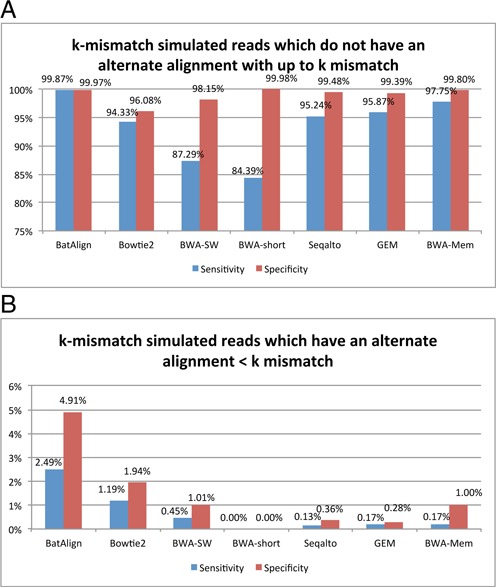
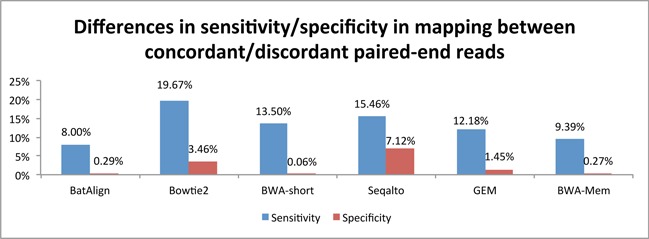
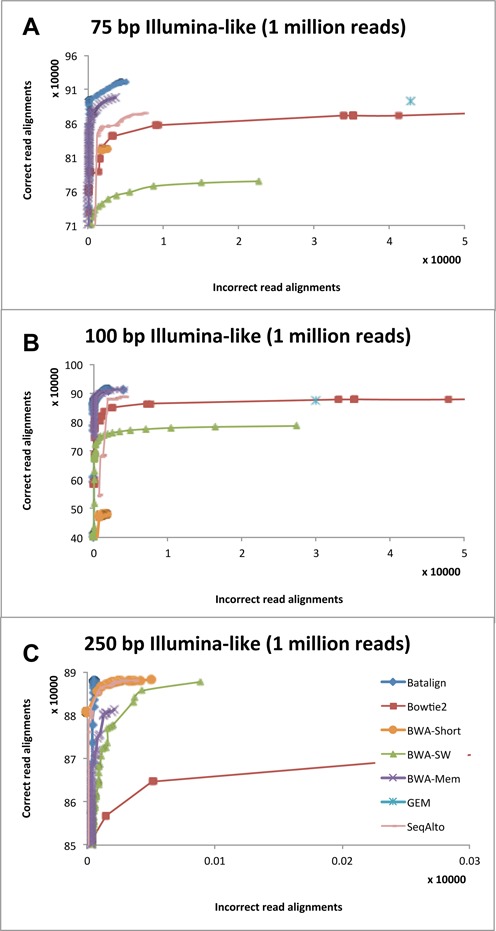
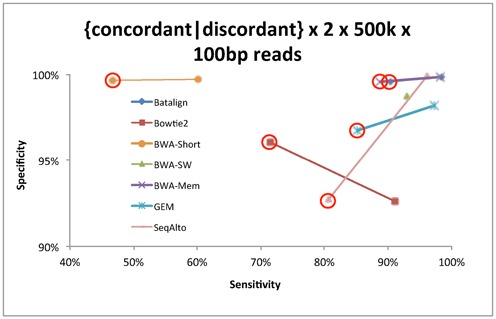
Structural variations (SVs) play a crucial role in genetic diversity. However, the alignments of reads near/across SVs are made inaccurate by the presence of polymorphisms. BatAlign is an algorithm that integrated two strategies called 'Reverse-Alignment' and 'Deep-Scan' to improve the accuracy of read-alignment. In our experiments, BatAlign was able to obtain the highest F-measures in read-alignments on mismatch-aberrant, indel-aberrant, concordantly/discordantly paired and SV-spanning data sets. On real data, the alignments of BatAlign were able to recover 4.3% more PCR-validated SVs with 73.3% less callings. These suggest BatAlign to be effective in detecting SVs and other polymorphic-variants accurately using high-throughput data. BatAlign is publicly available at https://goo.gl/a6phxB.
结构变异(SVs)在遗传多样性中起着至关重要的作用。然而,由于多态性的存在,SVs附近/跨越SVs的 reads 的比对变得不准确。BatAlign是一种整合了“反向比对”和“深度扫描”这两种策略的算法,以提高 reads 比对的准确性。在我们的实验中,BatAlign在错配异常、插入缺失异常、一致/不一致配对和跨越SVs的数据集的 reads 比对中能够获得最高的F值。在真实数据上,BatAlign的比对能够以少73.3%的调用次数多找回4.3%经PCR验证的SVs。这些表明BatAlign在使用高通量数据准确检测SVs和其他多态性变异方面是有效的。BatAlign可在https://goo.gl/a6phxB上公开获取。