Rukov Jakob Lewin, Hagedorn Peter H, Høy Isabel Bro, Feng Yanping, Lindow Morten, Vinther Jeppe
Department of Biology, University of Copenhagen, Ole Maaløes Vej 5, DK-2200 Copenhagen N, Denmark.
Roche Pharmaceutical Discovery and Early Development, Roche Innovation Center Copenhagen A/S, Fremtidsvej 3, DK-2970 Hørsholm, Denmark.
Nucleic Acids Res. 2015 Sep 30;43(17):8476-87. doi: 10.1093/nar/gkv759. Epub 2015 Jul 28.
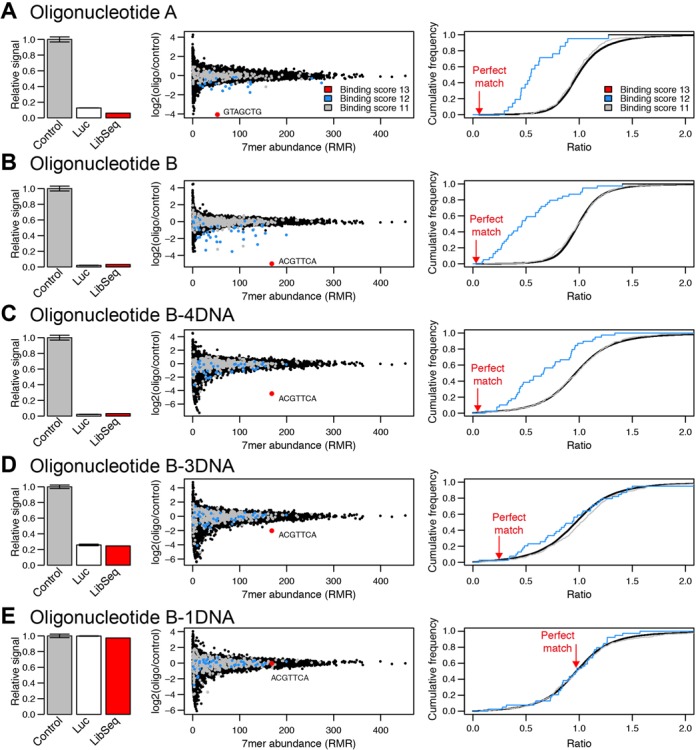
Processing and post-transcriptional regulation of RNA often depend on binding of regulatory molecules to short motifs in RNA. The effects of such interactions are difficult to study, because most regulatory molecules recognize partially degenerate RNA motifs, embedded in a sequence context specific for each RNA. Here, we describe Library Sequencing (LibSeq), an accurate massively parallel reporter method for completely characterizing the regulatory potential of thousands of short RNA sequences in a specific context. By sequencing cDNA derived from a plasmid library expressing identical reporter genes except for a degenerate 7mer subsequence in the 3'UTR, the regulatory effects of each 7mer can be determined. We show that LibSeq identifies regulatory motifs used by RNA-binding proteins and microRNAs. We furthermore apply the method to cells transfected with RNase H recruiting oligonucleotides to obtain quantitative information for >15000 potential target sequences in parallel. These comprehensive datasets provide insights into the specificity requirements of RNase H and allow a specificity measure to be calculated for each tested oligonucleotide. Moreover, we show that inclusion of chemical modifications in the central part of an RNase H recruiting oligonucleotide can increase its sequence-specificity.
RNA的加工和转录后调控通常依赖于调控分子与RNA中短基序的结合。此类相互作用的影响难以研究,因为大多数调控分子识别部分简并的RNA基序,这些基序嵌入在每个RNA特有的序列背景中。在此,我们描述了文库测序(LibSeq),这是一种准确的大规模平行报告基因方法,用于在特定背景下全面表征数千个短RNA序列的调控潜力。通过对来自质粒文库的cDNA进行测序,该文库除了3'UTR中的一个简并7聚体子序列外,表达相同的报告基因,从而可以确定每个7聚体的调控作用。我们表明,LibSeq可识别RNA结合蛋白和微小RNA使用的调控基序。我们还将该方法应用于转染了招募RNase H的寡核苷酸的细胞,以并行获取超过15000个潜在靶序列的定量信息。这些全面的数据集提供了对RNase H特异性要求的见解,并允许为每个测试的寡核苷酸计算特异性度量。此外,我们表明,在招募RNase H的寡核苷酸的中央部分引入化学修饰可以提高其序列特异性。