Lochovsky Lucas, Zhang Jing, Fu Yao, Khurana Ekta, Gerstein Mark
Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT 06520, USA.
Institute for Computational Biomedicine, Weill Cornell Medical College, New York, NY 10065, USA Department of Physiology and Biophysics, Weill Cornell Medical College, New York, New York 10065.
Nucleic Acids Res. 2015 Sep 30;43(17):8123-34. doi: 10.1093/nar/gkv803. Epub 2015 Aug 24.
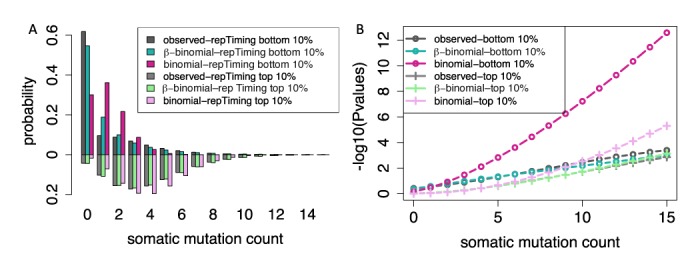
In cancer research, background models for mutation rates have been extensively calibrated in coding regions, leading to the identification of many driver genes, recurrently mutated more than expected. Noncoding regions are also associated with disease; however, background models for them have not been investigated in as much detail. This is partially due to limited noncoding functional annotation. Also, great mutation heterogeneity and potential correlations between neighboring sites give rise to substantial overdispersion in mutation count, resulting in problematic background rate estimation. Here, we address these issues with a new computational framework called LARVA. It integrates variants with a comprehensive set of noncoding functional elements, modeling the mutation counts of the elements with a β-binomial distribution to handle overdispersion. LARVA, moreover, uses regional genomic features such as replication timing to better estimate local mutation rates and mutational hotspots. We demonstrate LARVA's effectiveness on 760 whole-genome tumor sequences, showing that it identifies well-known noncoding drivers, such as mutations in the TERT promoter. Furthermore, LARVA highlights several novel highly mutated regulatory sites that could potentially be noncoding drivers. We make LARVA available as a software tool and release our highly mutated annotations as an online resource (larva.gersteinlab.org).
在癌症研究中,突变率的背景模型已在编码区得到广泛校准,从而识别出许多驱动基因,其突变频率反复高于预期。非编码区也与疾病相关;然而,针对它们的背景模型尚未得到如此详细的研究。部分原因是有限的非编码功能注释。此外,巨大的突变异质性以及相邻位点之间的潜在相关性导致突变计数出现大量过度离散,从而导致背景率估计存在问题。在此,我们使用一种名为LARVA的新计算框架来解决这些问题。它将变异与一套全面的非编码功能元件整合在一起,用β-二项分布对元件的突变计数进行建模以处理过度离散。此外,LARVA利用诸如复制时间等区域基因组特征来更好地估计局部突变率和突变热点。我们在760个全基因组肿瘤序列上证明了LARVA的有效性,表明它能识别出众所周知的非编码驱动因素,如TERT启动子中的突变。此外,LARVA突出了几个新的高度突变的调控位点,这些位点可能是潜在的非编码驱动因素。我们将LARVA作为一个软件工具提供,并将我们的高度突变注释作为在线资源发布(larva.gersteinlab.org)。