Yang Longhe, Li Long, Chen Ling, Li Yanting, Chen Huixia, Li Yuhang, Ji Guangnian, Lin Donghai, Liu Zuguo, Qiu Yan
Medical College, Xiamen University, Xiamen, Fujian 361005, P. R. China.
Engineering Research Center of Marine Biological Resource Comprehensive Utilization, Third Institute of Oceanography , State Oceanic Administration, Xiamen, Fujian 361005, P. R. China.
Sci Rep. 2015 Aug 27;5:13565. doi: 10.1038/srep13565.
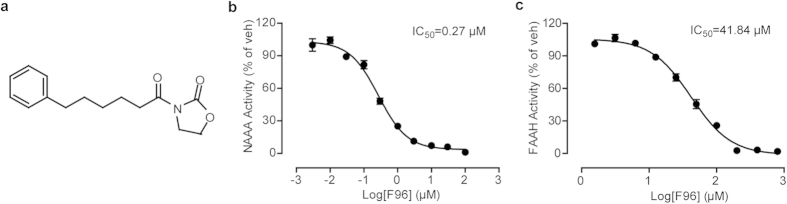
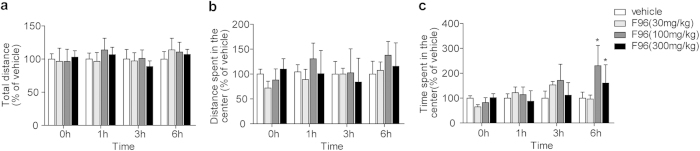
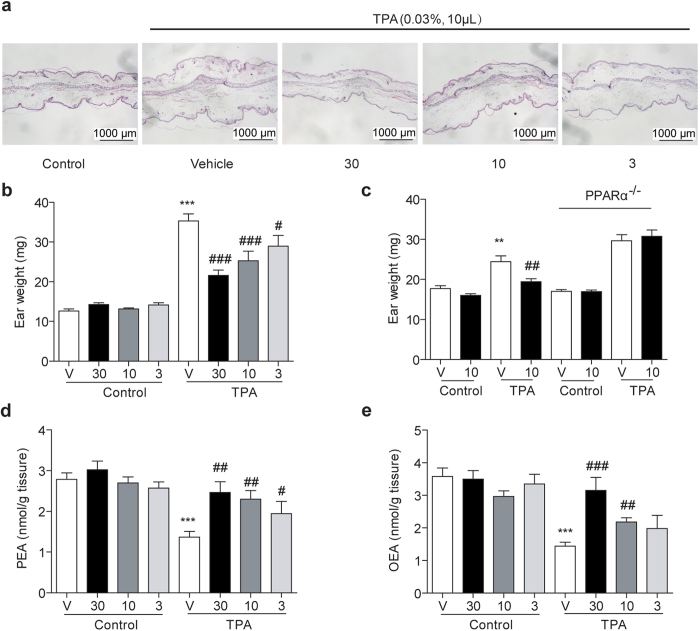
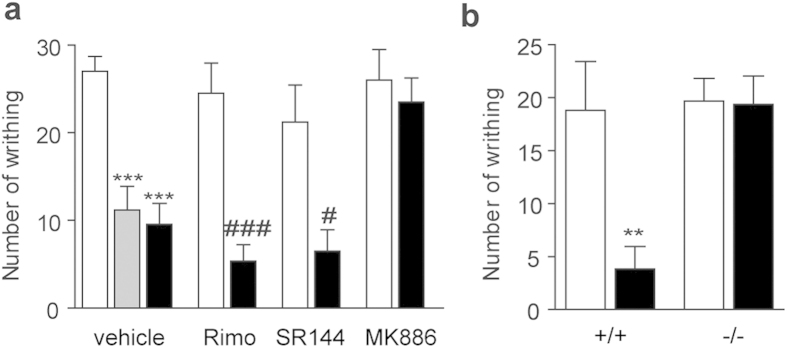
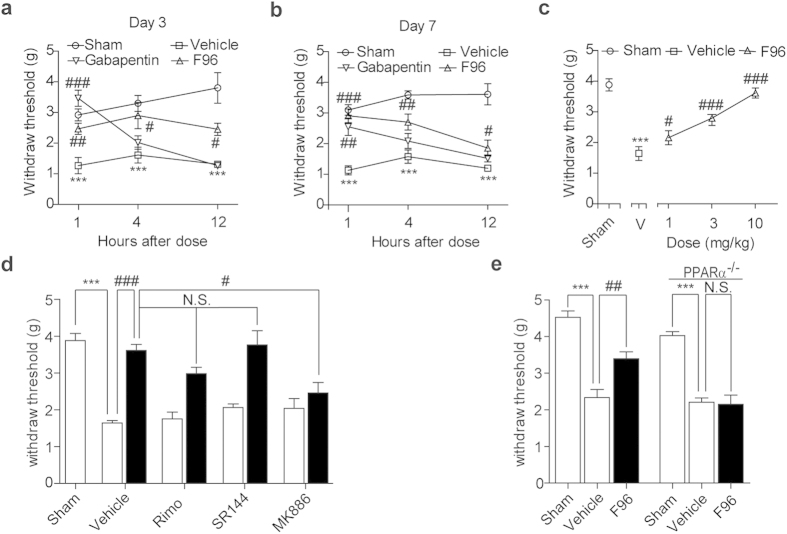
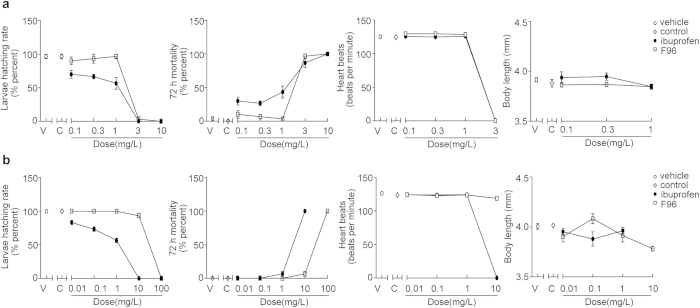
Pharmacological blockade of N-acylethanolamine acid amidase (NAAA) activity is an available approach for inflammation and pain control through restoring the ability of endogenous PEA. But the recently reported NAAA inhibitors suffer from the chemical and biological unstable properties, which restrict functions of NAAA inhibition in vivo. It is still unrevealed whether systematic inhibition of NAAA could modulate PEA-mediated pain signalings. Here we reported an oxazolidinone imide compound 3-(6-phenylhexanoyl) oxazolidin-2-one (F96), which potently and selectively inhibited NAAA activity (IC50 = 270 nM). Intraperitoneal (i.p.) injection of F96 (3-30 mg/kg) dose-dependently reduced ear edema and restored PEA levels of ear tissues in 12-O-Tetradecanoylphorbol-13-acetate (TPA) induced ear edema models. Furthermore, F96 inhibited acetic acid-induced writhing and increased spared nerve injury induced tactile allodynia thresholds in a dose-dependent manner. Pharmacological effects of F96 (10 mg/kg, i.p.) on various animal models were abolished in PPAR-α(-/-) mice, and were prevented by PPAR-α antagonist MK886 but not by canabinoid receptor type 1 (CB1) antagonist Rimonabant nor canabinoid receptor type 2 (CB2) antagonist SR144528. Zebrafish embryos experiments showed better security and lower toxicity for F96 than ibuprofen. These results revealed that F96 might be useful in treating inflammatory and neuropathic pain by NAAA inhibition depending on PPAR-α receptors.
通过恢复内源性棕榈酰乙醇胺(PEA)的能力,对N-酰基乙醇胺酸酰胺酶(NAAA)活性进行药理学阻断是控制炎症和疼痛的一种可行方法。但最近报道的NAAA抑制剂具有化学和生物学不稳定的特性,这限制了其在体内抑制NAAA的功能。目前仍不清楚系统性抑制NAAA是否能调节PEA介导的疼痛信号。在此,我们报道了一种恶唑烷酮酰亚胺化合物3-(6-苯基己酰基)恶唑烷-2-酮(F96),它能有效且选择性地抑制NAAA活性(IC50 = 270 nM)。在12-O-十四酰佛波醇-13-乙酸酯(TPA)诱导的耳部水肿模型中,腹腔注射F96(3 - 30 mg/kg)剂量依赖性地减轻耳部水肿,并恢复耳部组织中的PEA水平。此外,F96剂量依赖性地抑制乙酸诱导的扭体反应,并提高 spared nerve injury诱导的触觉异常性疼痛阈值。F96(10 mg/kg,腹腔注射)对各种动物模型的药理作用在PPAR-α(-/-)小鼠中被消除,并被PPAR-α拮抗剂MK886阻断,但未被大麻素受体-1(CB1)拮抗剂利莫那班和大麻素受体-2(CB2)拮抗剂SR144528阻断。斑马鱼胚胎实验表明,F96比布洛芬具有更好的安全性和更低的毒性。这些结果表明,F96可能通过依赖PPAR-α受体抑制NAAA来治疗炎症性和神经性疼痛。