Farrer Rhys A, Desjardins Christopher A, Sakthikumar Sharadha, Gujja Sharvari, Saif Sakina, Zeng Qiandong, Chen Yuan, Voelz Kerstin, Heitman Joseph, May Robin C, Fisher Matthew C, Cuomo Christina A
Genome Sequencing and Analysis Program, Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
Division of Infectious Diseases, Department of Medicine, Duke University Medical Center, Durham, North Carolina, USA.
mBio. 2015 Sep 1;6(5):e00868-15. doi: 10.1128/mBio.00868-15.
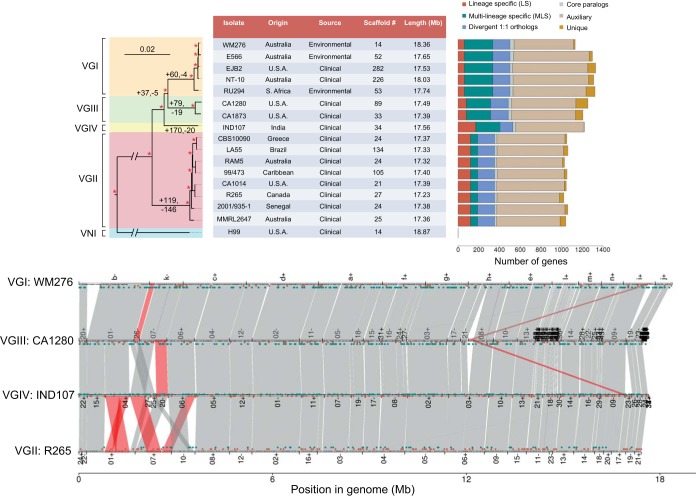
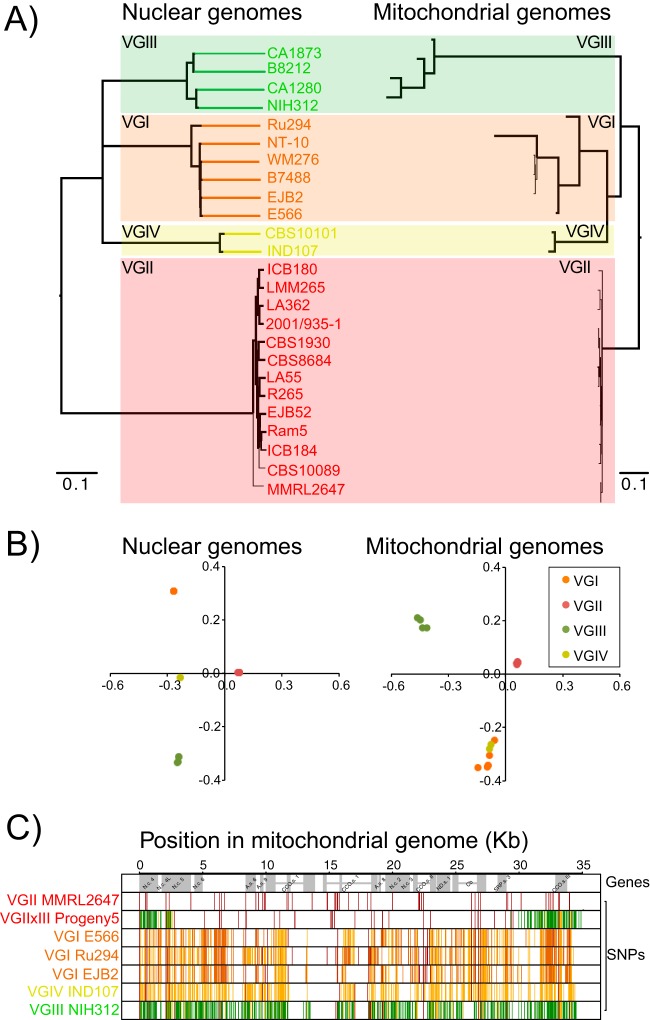
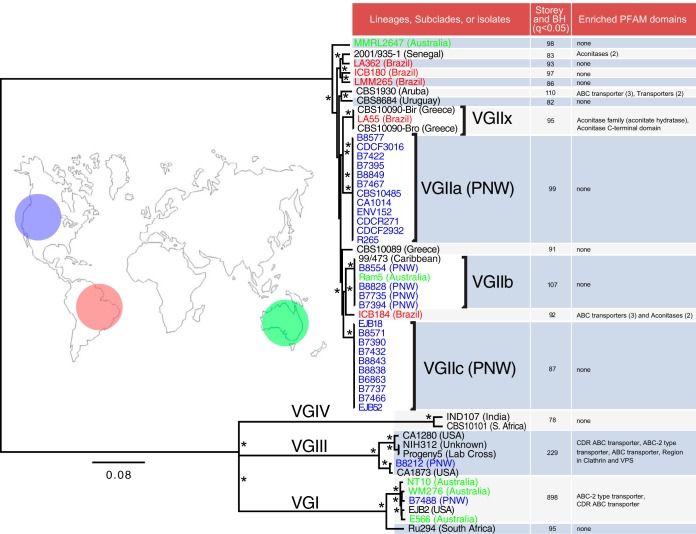
Cryptococcus gattii is a fungal pathogen of humans, causing pulmonary infections in otherwise healthy hosts. To characterize genomic variation among the four major lineages of C. gattii (VGI, -II, -III, and -IV), we generated, annotated, and compared 16 de novo genome assemblies, including the first for the rarely isolated lineages VGIII and VGIV. By identifying syntenic regions across assemblies, we found 15 structural rearrangements, which were almost exclusive to the VGI-III-IV lineages. Using synteny to inform orthology prediction, we identified a core set of 87% of C. gattii genes present as single copies in all four lineages. Remarkably, 737 genes are variably inherited across lineages and are overrepresented for response to oxidative stress, mitochondrial import, and metal binding and transport. Specifically, VGI has an expanded set of iron-binding genes thought to be important to the virulence of Cryptococcus, while VGII has expansions in the stress-related heat shock proteins relative to the other lineages. We also characterized genes uniquely absent in each lineage, including a copper transporter absent from VGIV, which influences Cryptococcus survival during pulmonary infection and the onset of meningoencephalitis. Through inclusion of population-level data for an additional 37 isolates, we identified a new transcontinental clonal group that we name VGIIx, mitochondrial recombination between VGII and VGIII, and positive selection of multidrug transporters and the iron-sulfur protein aconitase along multiple branches of the phylogenetic tree. Our results suggest that gene expansion or contraction and positive selection have introduced substantial variation with links to mechanisms of pathogenicity across this species complex.
The genetic differences between phenotypically different pathogens provide clues to the underlying mechanisms of those traits and can lead to new drug targets and improved treatments for those diseases. In this paper, we compare 16 genomes belonging to four highly differentiated lineages of Cryptococcus gattii, which cause pulmonary infections in otherwise healthy humans and other animals. Half of these lineages have not had their genomes previously assembled and annotated. We identified 15 ancestral rearrangements in the genome and over 700 genes that are unique to one or more lineages, many of which are associated with virulence. In addition, we found evidence for recent transcontinental spread, mitochondrial genetic exchange, and positive selection in multidrug transporters. Our results suggest that gene expansion/contraction and positive selection are diversifying the mechanisms of pathogenicity across this species complex.
加氏隐球菌是一种人类真菌病原体,可在原本健康的宿主中引起肺部感染。为了表征加氏隐球菌四个主要谱系(VGI、-II、-III和-IV)之间的基因组变异,我们生成、注释并比较了16个从头基因组组装序列,包括首次对罕见分离的VGIII和VGIV谱系进行的组装。通过识别各组装序列间的同线性区域,我们发现了15个结构重排,这些重排几乎是VGI-III-IV谱系所特有的。利用同线性来指导直系同源预测,我们确定了一组核心基因,在所有四个谱系中,87%的加氏隐球菌基因以单拷贝形式存在。值得注意的是,737个基因在各谱系间的遗传方式存在差异,且在氧化应激反应、线粒体导入以及金属结合与转运方面显著富集。具体而言,VGI具有一组扩增的铁结合基因,这些基因被认为对隐球菌的毒力很重要,而相对于其他谱系,VGII在与应激相关的热休克蛋白方面有所扩增。我们还表征了每个谱系中独特缺失的基因,包括VGIV中缺失的一种铜转运蛋白,该蛋白影响肺部感染期间隐球菌的存活以及脑膜脑炎的发病。通过纳入另外37个分离株的群体水平数据,我们确定了一个新的跨大陆克隆群,命名为VGIIx,发现了VGII和VGIII之间的线粒体重组,以及在系统发育树的多个分支上多药转运蛋白和铁硫蛋白乌头酸酶的正选择。我们的结果表明,基因扩增或收缩以及正选择在这个物种复合体中引入了与致病机制相关的大量变异。
表型不同的病原体之间的遗传差异为这些性状的潜在机制提供了线索,并可导致新的药物靶点和针对这些疾病的改进治疗方法。在本文中,我们比较了属于加氏隐球菌四个高度分化谱系的16个基因组,这些谱系可在原本健康的人类和其他动物中引起肺部感染。其中一半的谱系此前尚未进行基因组组装和注释。我们在基因组中鉴定出15个祖先重排以及700多个特定于一个或多个谱系的基因,其中许多与毒力相关。此外,我们发现了近期跨大陆传播、线粒体基因交换以及多药转运蛋白正选择的证据。我们的结果表明,基因扩增/收缩和正选择正在使这个物种复合体的致病机制多样化。