Wen Zixiang, Boyse John F, Song Qijian, Cregan Perry B, Wang Dechun
Department of Plant, Soil and Microbial Sciences, Michigan State University, 1066 Bogue St., Rm. A384-E, East Lansing, MI, 48824-1325, USA.
Soybean Genomics and Improvement Laboratory, Agricultural Research Service, United States Department of Agriculture, Beltsville, MD, 20705, USA.
BMC Genomics. 2015 Sep 3;16(1):671. doi: 10.1186/s12864-015-1872-y.
Crop improvement always involves selection of specific alleles at genes controlling traits of agronomic importance, likely resulting in detectable signatures of selection within the genome of modern soybean (Glycine max L. Merr.). The identification of these signatures of selection is meaningful from the perspective of evolutionary biology and for uncovering the genetic architecture of agronomic traits.
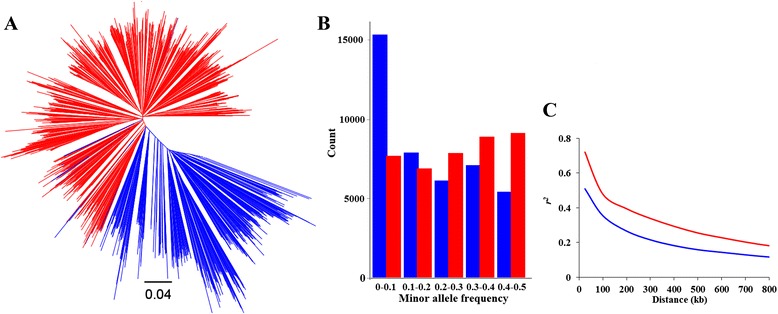
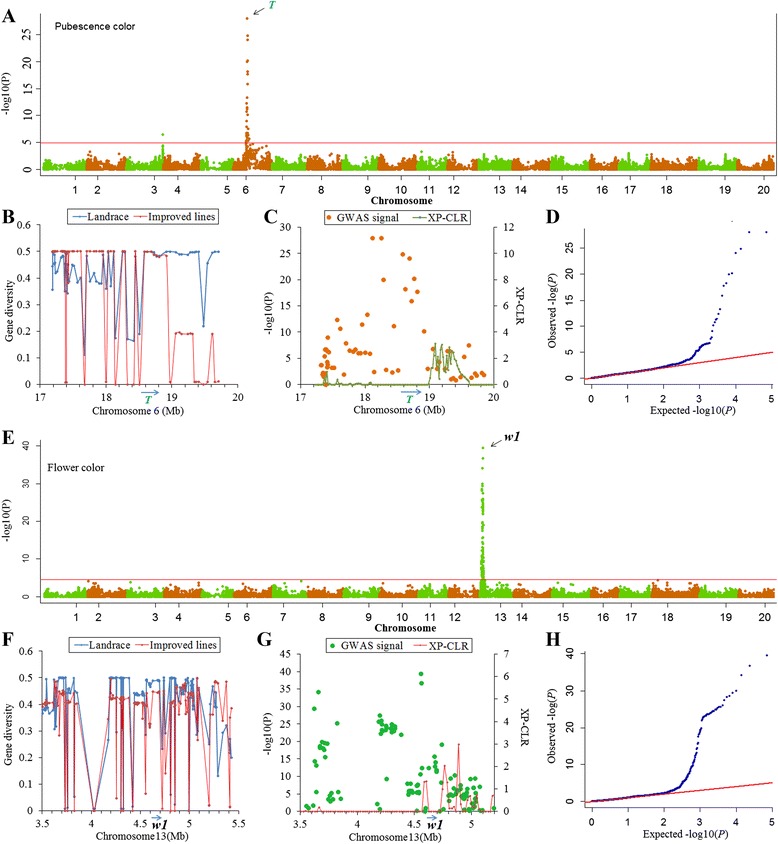
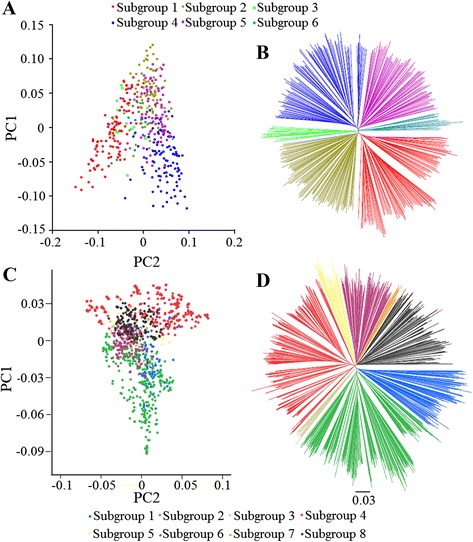
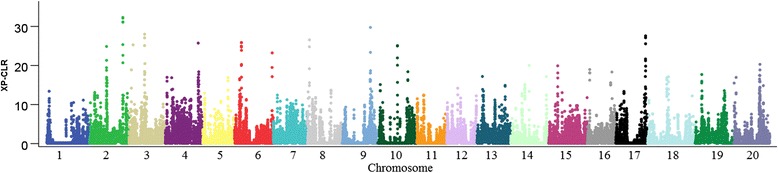
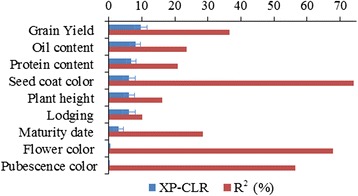
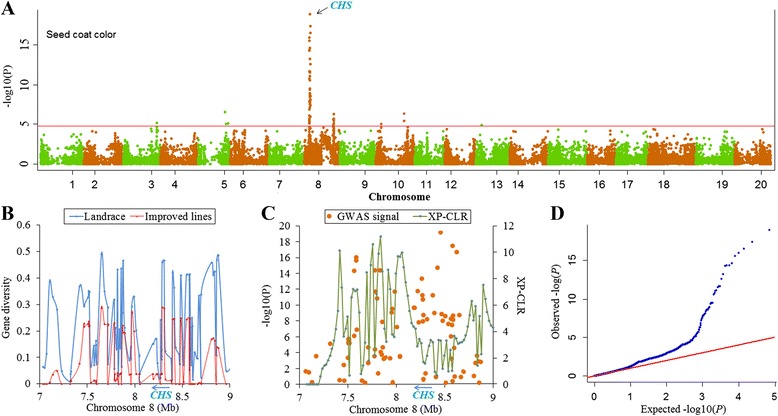
To this end, two populations of soybean, consisting of 342 landraces and 1062 improved lines, were genotyped with the SoySNP50K Illumina BeadChip containing 52,041 single nucleotide polymorphisms (SNPs), and systematically phenotyped for 9 agronomic traits. A cross-population composite likelihood ratio (XP-CLR) method was used to screen the signals of selective sweeps. A total of 125 candidate selection regions were identified, many of which harbored genes potentially involved in crop improvement. To further investigate whether these candidate regions were in fact enriched for genes affected by selection, genome-wide association studies (GWAS) were conducted on 7 selection traits targeted in soybean breeding (grain yield, plant height, lodging, maturity date, seed coat color, seed protein and oil content) and 2 non-selection traits (pubescence and flower color). Major genomic regions associated with selection traits overlapped with candidate selection regions, whereas no overlap of this kind occurred for the non-selection traits, suggesting that the selection sweeps identified are associated with traits of agronomic importance. Multiple novel loci and refined map locations of known loci related to these traits were also identified.
These findings illustrate that comparative genomic analyses, especially when combined with GWAS, are a promising approach to dissect the genetic architecture of complex traits.
作物改良总是涉及在控制重要农艺性状的基因处选择特定等位基因,这可能会在现代大豆(Glycine max L. Merr.)基因组中产生可检测到的选择印记。从进化生物学角度以及揭示农艺性状的遗传结构来看,识别这些选择印记具有重要意义。
为此,对两个大豆群体(342份地方品种和1062份改良品系)进行了基因分型,使用包含52,041个单核苷酸多态性(SNP)的SoySNP50K Illumina BeadChip芯片,并对9个农艺性状进行了系统表型分析。采用跨群体复合似然比(XP-CLR)方法筛选选择清除信号。共鉴定出125个候选选择区域,其中许多区域含有可能参与作物改良的基因。为了进一步研究这些候选区域是否实际上富集了受选择影响的基因,对大豆育种中针对的7个选择性状(籽粒产量、株高、倒伏性、成熟日期、种皮颜色、种子蛋白质和油含量)和2个非选择性状(茸毛和花色)进行了全基因组关联研究(GWAS)。与选择性状相关的主要基因组区域与候选选择区域重叠,而非选择性状则未出现这种重叠,这表明所识别的选择清除与重要农艺性状相关。还鉴定出了与这些性状相关的多个新位点以及已知位点的精细图谱位置。
这些发现表明,比较基因组分析,尤其是与GWAS相结合时,是剖析复杂性状遗传结构的一种有前途的方法。