Soto Angela Ganan, McIntyre Adam, Agrawal Sungeeta, Bialo Shara R, Hegele Robert A, Boney Charlotte M
Department of Pediatrics, Rhode Island Hospital and Brown University, Providence, RI, USA.
Robarts Research Institute, Western University, London, ON, Canada.
Lipids Health Dis. 2015 Sep 4;14:102. doi: 10.1186/s12944-015-0107-1.
Lipoprotein Lipase (LPL) deficiency is a rare autosomal recessive disorder with a heterogeneous clinical presentation. Several mutations in the LPL gene have been identified to cause decreased activity of the enzyme.
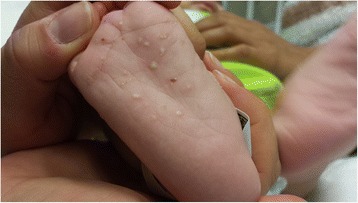
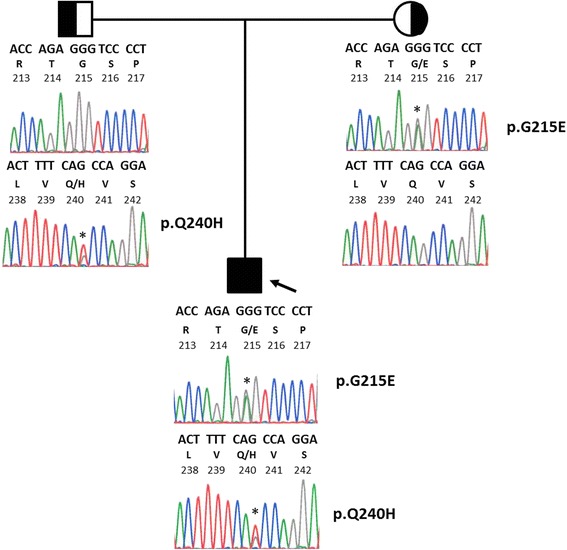
An 11-week-old, exclusively breastfed male presented with coffee-ground emesis, melena, xanthomas, lipemia retinalis and chylomicronemia. Genomic DNA analysis identified lipoprotein lipase deficiency due to compound heterozygosity including a novel p.Q240H mutation in exon 5 of the lipoprotein lipase (LPL) gene. His severe hypertriglyceridemia, including xanthomas, resolved with dietary long-chain fat restriction.
We describe a novel mutation of the LPL gene causing severe hypertriglyceridemia and report the response to treatment. A review of the current literature regarding LPL deficiency syndrome reveals a few potential new therapies under investigation.
脂蛋白脂肪酶(LPL)缺乏症是一种罕见的常染色体隐性疾病,临床表现具有异质性。已确定LPL基因中的几种突变会导致该酶活性降低。
一名11周大、纯母乳喂养的男性出现咖啡渣样呕吐物、黑便、黄色瘤、视网膜脂血症和乳糜微粒血症。基因组DNA分析确定由于复合杂合性导致脂蛋白脂肪酶缺乏,包括脂蛋白脂肪酶(LPL)基因第5外显子中的一个新的p.Q240H突变。他的严重高甘油三酯血症,包括黄色瘤,通过饮食中长链脂肪限制得到缓解。
我们描述了一种导致严重高甘油三酯血症的LPL基因新突变,并报告了治疗反应。对目前关于LPL缺乏综合征的文献综述显示,有几种潜在的新疗法正在研究中。