Lebeaupin C, Proics E, de Bieville C H D, Rousseau D, Bonnafous S, Patouraux S, Adam G, Lavallard V J, Rovere C, Le Thuc O, Saint-Paul M C, Anty R, Schneck A S, Iannelli A, Gugenheim J, Tran A, Gual P, Bailly-Maitre B
INSERM, U1065, Equipe 8 « Complications hépatiques de l'obésité », Bâtiment Universitaire ARCHIMED, Nice, France.
Université de Nice Sophia Antipolis, Faculté de Médecine, Nice, France.
Cell Death Dis. 2015 Sep 10;6(9):e1879. doi: 10.1038/cddis.2015.248.
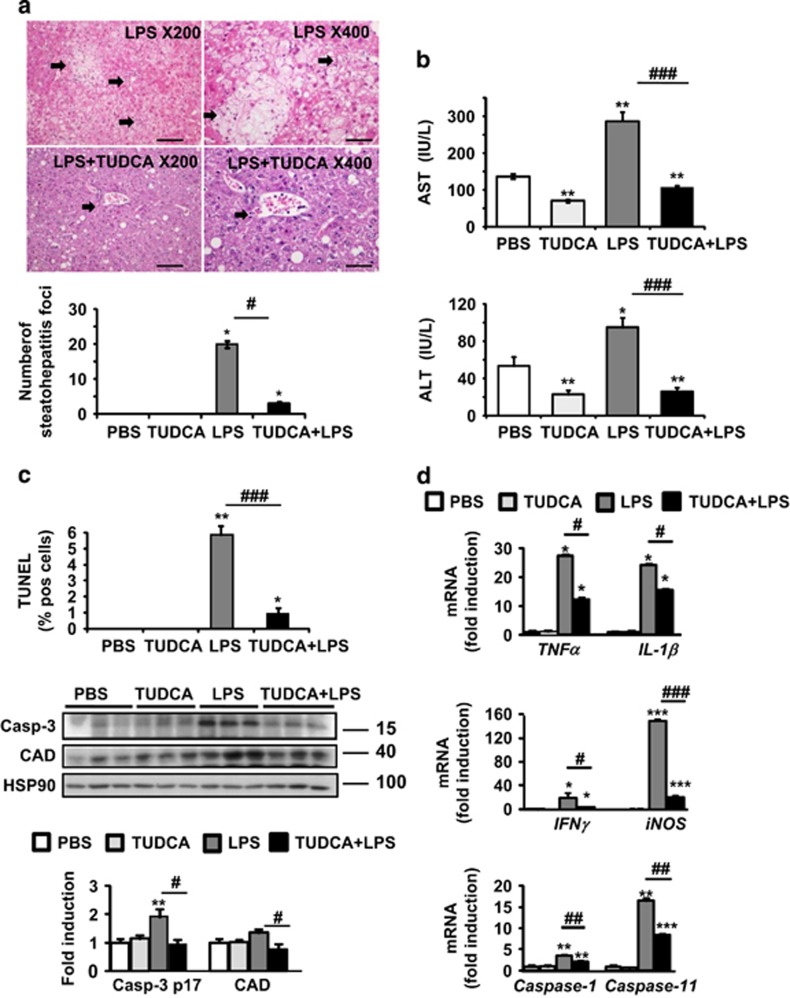
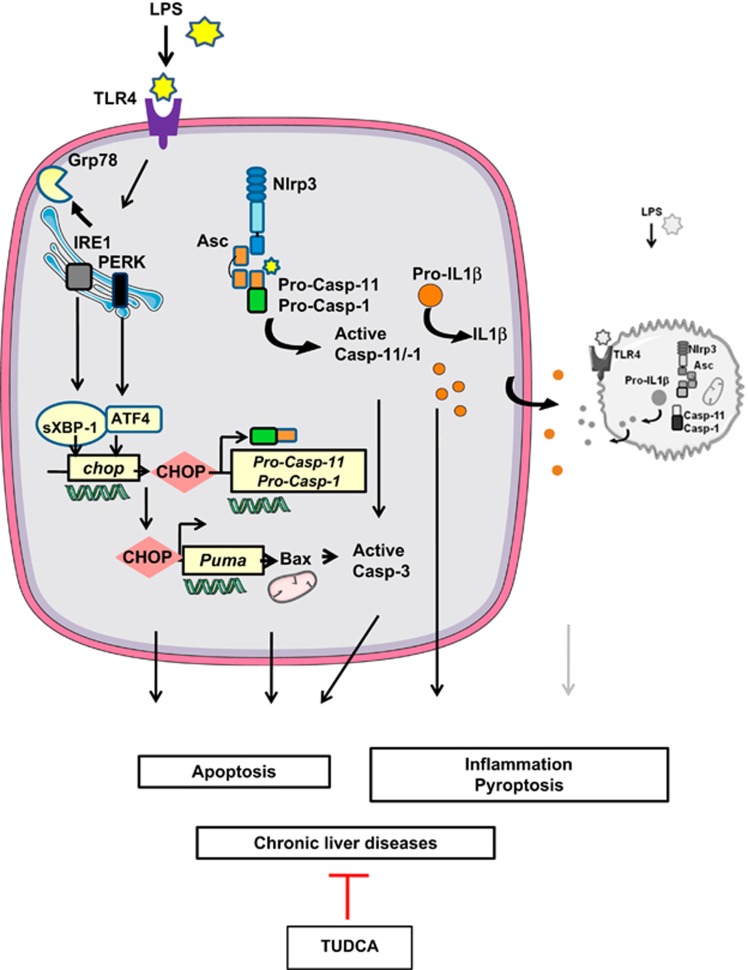
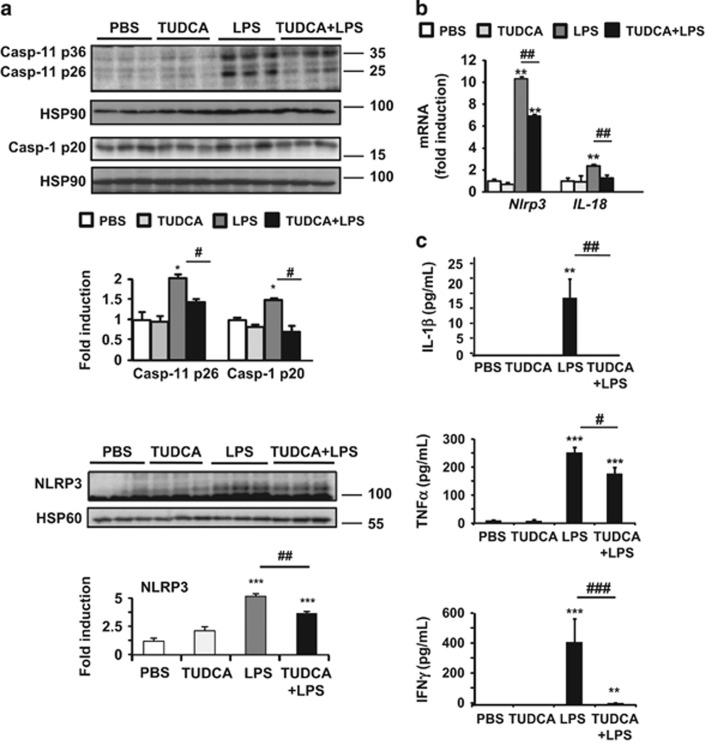
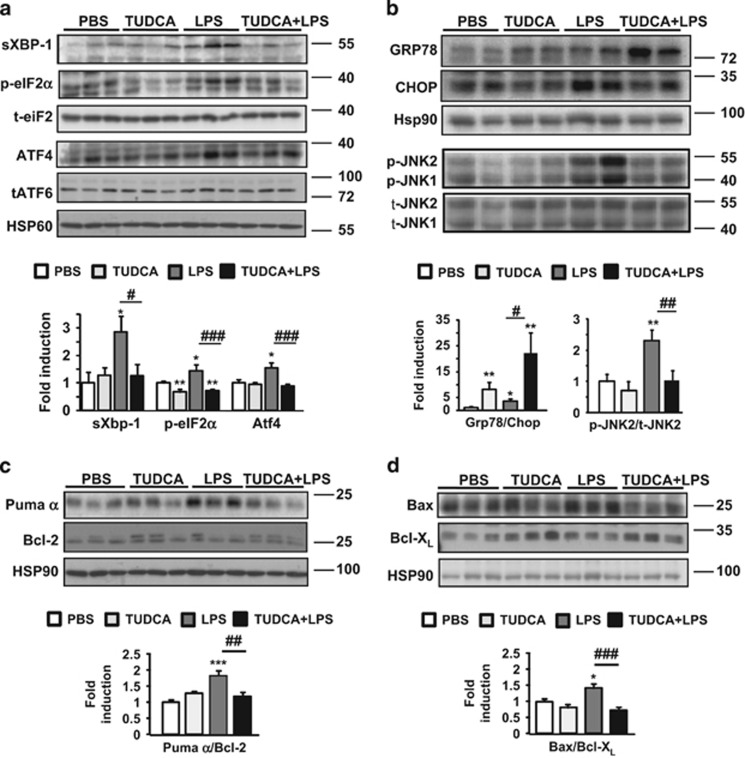
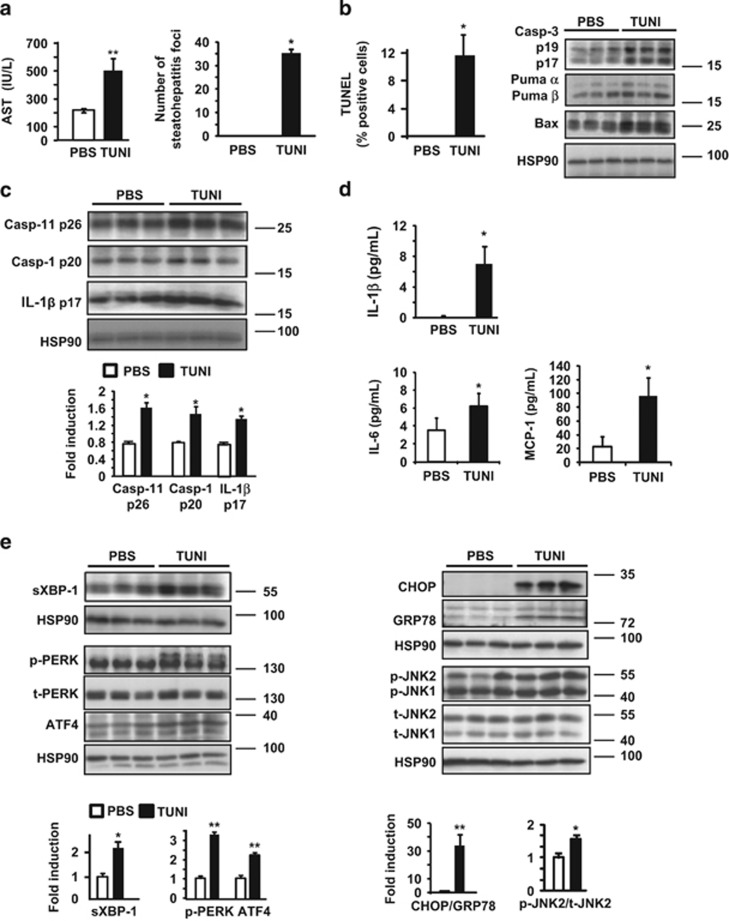
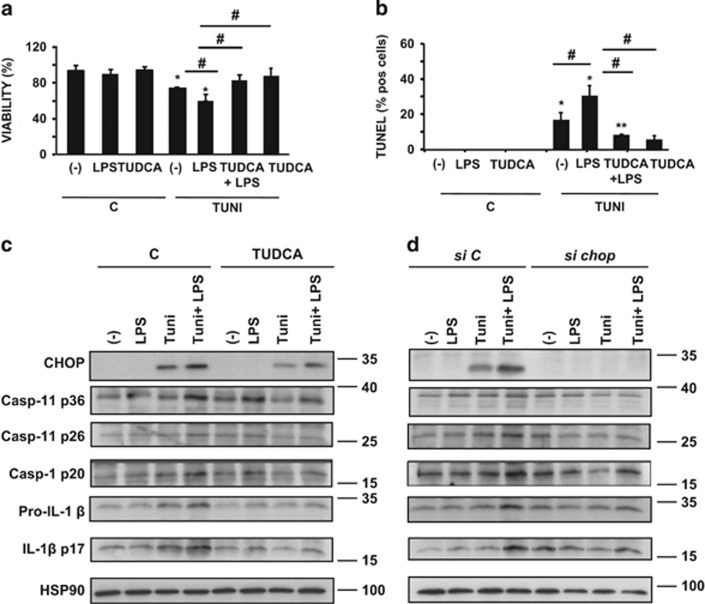
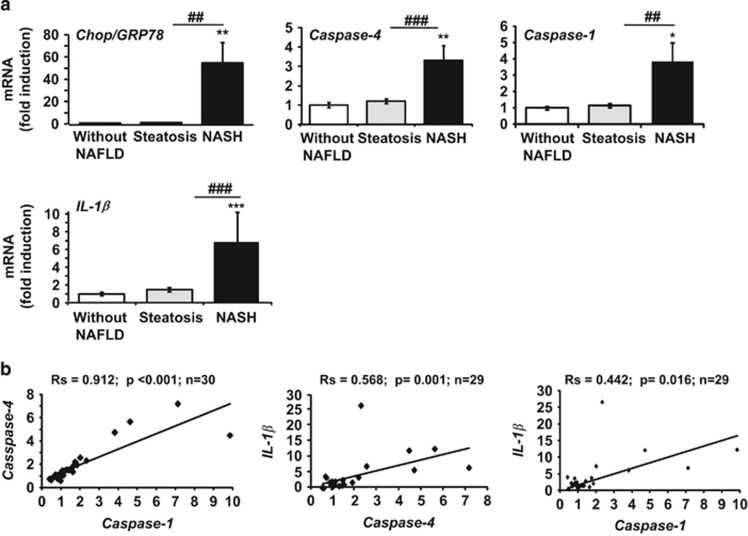
The incidence of chronic liver disease is constantly increasing, owing to the obesity epidemic. However, the causes and mechanisms of inflammation-mediated liver damage remain poorly understood. Endoplasmic reticulum (ER) stress is an initiator of cell death and inflammatory mechanisms. Although obesity induces ER stress, the interplay between hepatic ER stress, NLRP3 inflammasome activation and hepatocyte death signaling has not yet been explored during the etiology of chronic liver diseases. Steatosis is a common disorder affecting obese patients; moreover, 25% of these patients develop steatohepatitis with an inherent risk for progression to hepatocarcinoma. Increased plasma LPS levels have been detected in the serum of patients with steatohepatitis. We hypothesized that, as a consequence of increased plasma LPS, ER stress could be induced and lead to NLRP3 inflammasome activation and hepatocyte death associated with steatohepatitis progression. In livers from obese mice, administration of LPS or tunicamycin results in IRE1α and PERK activation, leading to the overexpression of CHOP. This, in turn, activates the NLRP3 inflammasome, subsequently initiating hepatocyte pyroptosis (caspase-1, -11, interleukin-1β secretion) and apoptosis (caspase-3, BH3-only proteins). In contrast, the LPS challenge is blocked by the ER stress inhibitor TUDCA, resulting in: CHOP downregulation, reduced caspase-1, caspase-11, caspase-3 activities, lowered interleukin-1β secretion and rescue from cell death. The central role of CHOP in mediating the activation of proinflammatory caspases and cell death was characterized by performing knockdown experiments in primary mouse hepatocytes. Finally, the analysis of human steatohepatitis liver biopsies showed a correlation between the upregulation of inflammasome and ER stress markers, as well as liver injury. We demonstrate here that ER stress leads to hepatic NLRP3 inflammasome pyroptotic death, thus contributing as a novel mechanism of inflammation-mediated liver injury in chronic liver diseases. Inhibition of ER-dependent inflammasome activation and cell death pathways may represent a potential therapeutic approach in chronic liver diseases.
由于肥胖症的流行,慢性肝病的发病率正在不断上升。然而,炎症介导的肝损伤的原因和机制仍知之甚少。内质网(ER)应激是细胞死亡和炎症机制的启动因素。虽然肥胖会诱导内质网应激,但在慢性肝病的病因学中,肝脏内质网应激、NLRP3炎性小体激活和肝细胞死亡信号之间的相互作用尚未得到探索。脂肪变性是影响肥胖患者的常见病症;此外,这些患者中有25%会发展为脂肪性肝炎,并有进展为肝癌的内在风险。在脂肪性肝炎患者的血清中检测到血浆LPS水平升高。我们假设,由于血浆LPS增加,内质网应激可能被诱导,并导致NLRP3炎性小体激活和与脂肪性肝炎进展相关的肝细胞死亡。在肥胖小鼠的肝脏中,给予LPS或衣霉素会导致IRE1α和PERK激活,从而导致CHOP的过度表达。这反过来又会激活NLRP3炎性小体,随后引发肝细胞焦亡(半胱天冬酶-1、-11、白细胞介素-1β分泌)和凋亡(半胱天冬酶-3、仅含BH3结构域的蛋白质)。相反,内质网应激抑制剂TUDCA可阻断LPS刺激,导致:CHOP下调、半胱天冬酶-1、半胱天冬酶-11、半胱天冬酶-3活性降低、白细胞介素-1β分泌减少,并使细胞免于死亡。通过在原代小鼠肝细胞中进行敲低实验,确定了CHOP在介导促炎性半胱天冬酶激活和细胞死亡中的核心作用。最后,对人类脂肪性肝炎肝活检的分析表明,炎性小体和内质网应激标志物的上调与肝损伤之间存在相关性。我们在此证明内质网应激会导致肝脏NLRP3炎性小体焦亡死亡,从而作为慢性肝病中炎症介导的肝损伤的一种新机制。抑制内质网依赖性炎性小体激活和细胞死亡途径可能代表了慢性肝病的一种潜在治疗方法。