Vandervalk Benjamin P, Yang Chen, Xue Zhuyi, Raghavan Karthika, Chu Justin, Mohamadi Hamid, Jackman Shaun D, Chiu Readman, Warren René L, Birol Inanç
BMC Med Genomics. 2015;8 Suppl 3(Suppl 3):S1. doi: 10.1186/1755-8794-8-S3-S1. Epub 2015 Sep 23.
Reading the nucleotides from two ends of a DNA fragment is called paired-end tag (PET) sequencing. When the fragment length is longer than the combined read length, there remains a gap of unsequenced nucleotides between read pairs. If the target in such experiments is sequenced at a level to provide redundant coverage, it may be possible to bridge these gaps using bioinformatics methods. Konnector is a local de novo assembly tool that addresses this problem. Here we report on version 2.0 of our tool.
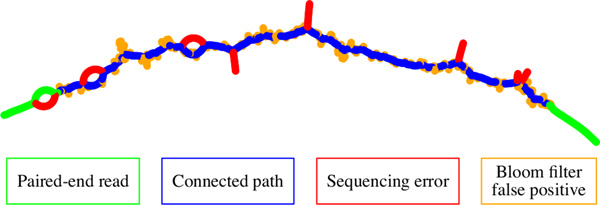
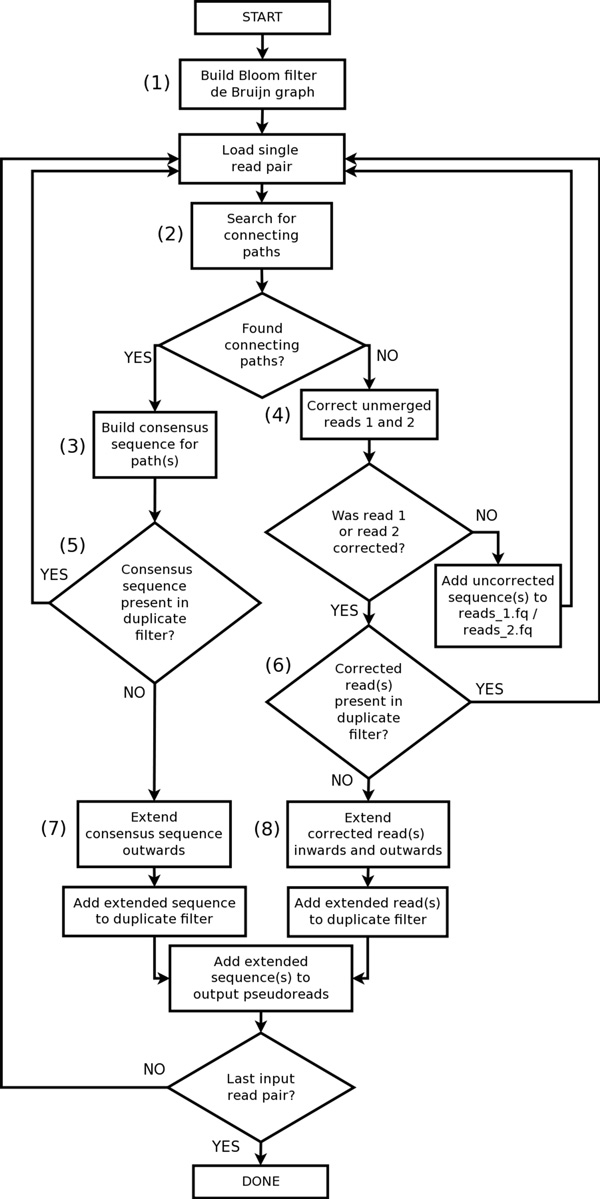
Konnector uses a probabilistic and memory-efficient data structure called Bloom filter to represent a k-mer spectrum - all possible sequences of length k in an input file, such as the collection of reads in a PET sequencing experiment. It performs look-ups to this data structure to construct an implicit de Bruijn graph, which describes (k-1) base pair overlaps between adjacent k-mers. It traverses this graph to bridge the gap between a given pair of flanking sequences.
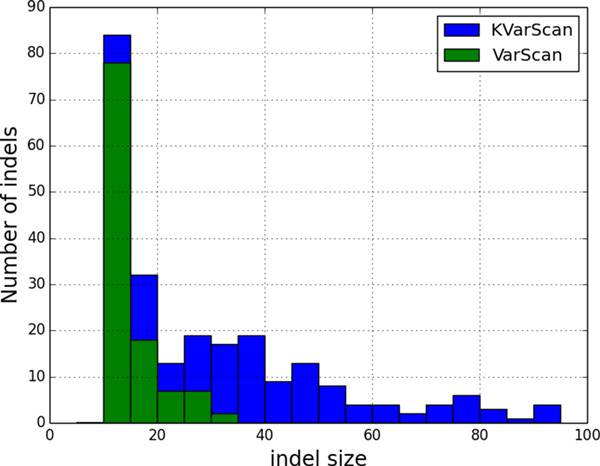
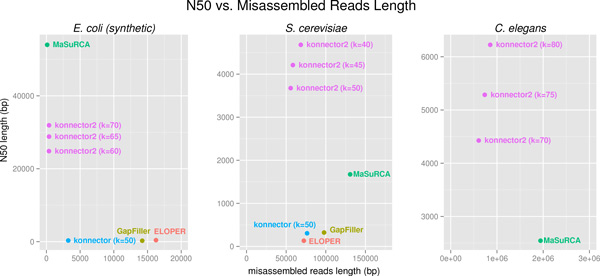
Here we report the performance of Konnector v2.0 on simulated and experimental datasets, and compare it against other tools with similar functionality. We note that, representing k-mers with 1.5 bytes of memory on average, Konnector can scale to very large genomes. With our parallel implementation, it can also process over a billion bases on commodity hardware.
从DNA片段的两端读取核苷酸被称为双末端标签(PET)测序。当片段长度长于组合读取长度时,读取对之间会存在未测序核苷酸的间隙。如果此类实验中的目标测序达到提供冗余覆盖的水平,那么使用生物信息学方法有可能填补这些间隙。Konnector是一种解决此问题的本地从头组装工具。在此我们报告该工具的2.0版本。
Konnector使用一种名为布隆过滤器的概率性且内存高效的数据结构来表示k-mer频谱——输入文件中所有长度为k的可能序列,例如PET测序实验中的读取集合。它对该数据结构进行查找以构建一个隐式德布鲁因图,该图描述相邻k-mer之间的(k-1)碱基对重叠。它遍历此图以填补给定一对侧翼序列之间的间隙。
在此我们报告Konnector v2.0在模拟和实验数据集上的性能,并将其与其他具有类似功能的工具进行比较。我们注意到,Konnector平均用1.5字节内存表示k-mer,能够扩展到非常大的基因组。通过我们的并行实现,它还能在商用硬件上处理超过十亿个碱基。