Jabbehdari Sayena, Rahimian Elham, Jafari Narjes, Sanii Sara, Khayatzadehkakhki Simin, Nejad Biglari Habibe
Students' Research Committee, Faculty of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran.
Neuroradiologist, Haghighat Radiology Center, Tehran, Iran.
Iran J Child Neurol. 2015 Summer;9(3):57-61.
Metachromatic leukodystrophy disorder (MLD) is one of the rare neurometabolic diseases caused due to lack of saposin B and arylsulfatase A enzyme deficiency.
MATERIALS & METHODS: Eighteen patients diagnosed as metachromatic leukodystrophy in the Neurology Department of Mofid Children's Hospital in Tehran, Iran between 2010 and 2014 were included in our study. The disorder was confirmed by clinical, EMG-NCV, arylsulfatase A enzyme checking and neuroimaging findings along with neurometabolic and genetic assessment from reference laboratory in Iran. We assessed age, gender, past medical history, developmental status, clinical manifestations, and neuroimaging findings of 18 patients with metachromatic leukodystrophy.
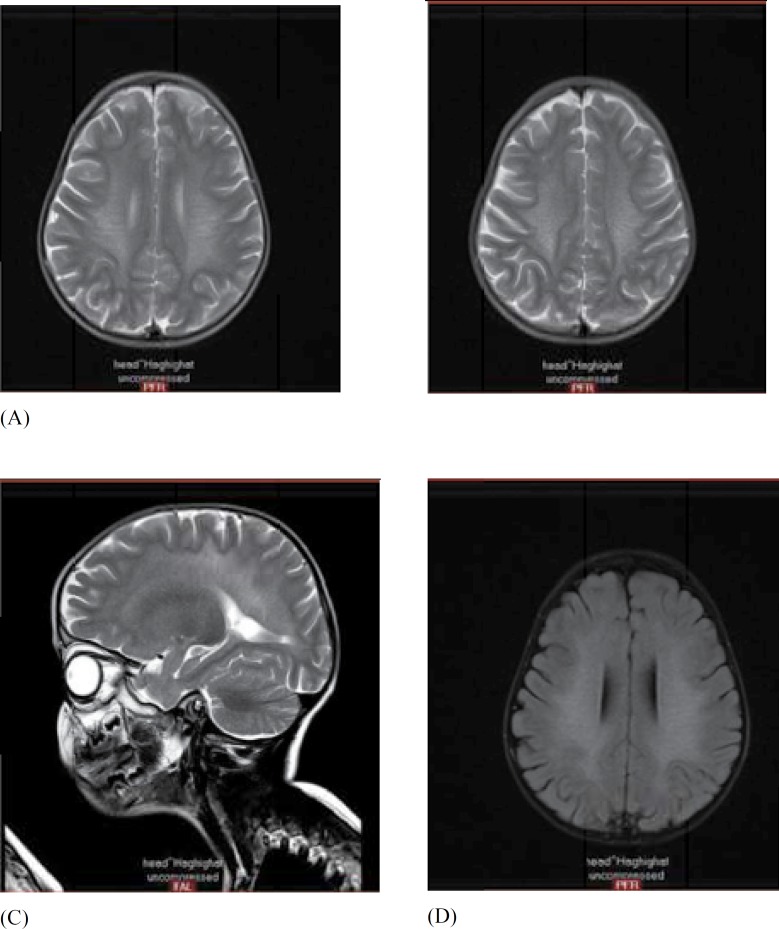
From 18 patients, 80% were offspring from consanguineous marriages. A family history of metachromatic leukodystrophy disease was positive for four patients. Twelve patients had late infantile form of this disorder and six patients had juvenile form. A history of tonic type seizure was positive in 20% of the patients and tonic spasm was confirmed with clinical information. Electromyographgraphy (EMG) in 96% of patients was abnormal with demyelinating sensorimotor neuropathy pattern. MRI in all patients showed the leukodystrophic pattern as arcuate fibers sparing and subcortical rim in white matter and periventricular involvement. Our diagnosis was confirmed by EMG-NCV findings with sensorimotor neuropathy pattern and the assessment of arylsulfatase A enzyme function.
MLD is an inheritance metabolic disorder, which was confirmed by the assessment of arylsulfatase A enzyme function, peripheral blood leukocyte that assessed in a referral laboratory in Iran.
异染性脑白质营养不良症(MLD)是一种罕见的神经代谢疾病,由缺乏鞘脂激活蛋白B和芳基硫酸酯酶A酶缺乏引起。
纳入2010年至2014年在伊朗德黑兰莫菲德儿童医院神经科诊断为异染性脑白质营养不良的18例患者。通过临床、肌电图-神经传导速度检查、芳基硫酸酯酶A酶检测和神经影像学检查结果,以及来自伊朗参考实验室的神经代谢和基因评估,确诊该疾病。我们评估了18例异染性脑白质营养不良患者的年龄、性别、既往病史、发育状况、临床表现和神经影像学检查结果。
18例患者中,80%为近亲结婚的后代。4例患者有家族性异染性脑白质营养不良病史。12例患者为该疾病的晚婴型,6例患者为青少年型。20%的患者有强直性癫痫病史,临床信息证实有强直性痉挛。96%的患者肌电图(EMG)异常,呈脱髓鞘感觉运动神经病模式。所有患者的MRI均显示脑白质营养不良模式,即弓形纤维保留、白质皮质下边缘和脑室周围受累。我们的诊断通过EMG-NCV检查结果呈感觉运动神经病模式以及芳基硫酸酯酶A酶功能评估得到证实。
MLD是一种遗传性代谢疾病,通过在伊朗一家转诊实验室评估芳基硫酸酯酶A酶功能和外周血白细胞得以证实。