Bradley William H, Eng Kevin, Le Min, Mackinnon A Craig, Kendziorski Christina, Rader Janet S
Department of Obstetrics and Gynecology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226 USA.
Department of Biostatistics and Medical Informatics, University of Wisconsin-Madison, Madison, WI 53792 USA ; Current Address: Department of Biostatistics and Bioinformatics, Roswell Park Cancer Institute, Buffalo, NY USA.
BMC Clin Pathol. 2015 Sep 24;15:17. doi: 10.1186/s12907-015-0017-1. eCollection 2015.
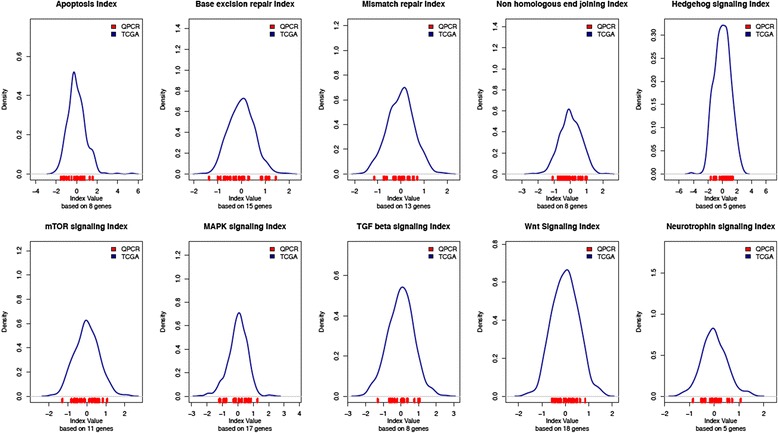
Previously, we have used clinical and gene expression data from The Cancer Genome Atlas (TCGA) to model a pathway-based index predicting outcomes in ovarian carcinoma. This data were obtained from snap-frozen tissue measured with the Affymetrix U133 platform. In the current study, we correlate the data used to model with data derived from TaqMan qPCR both snap frozen and paraffin embedded (FFPE) samples.
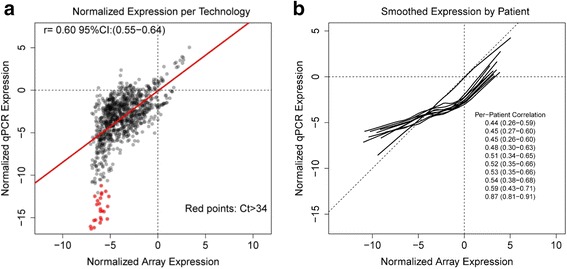
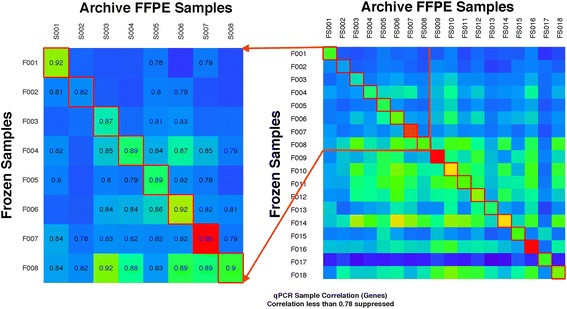
To compare the effect of preservation methods on gene expression measured by qPCR, we assessed 18 patient and tumor sample matched snap-frozen and FFPE ovarian carcinoma samples. To compare gene measurement technologies, we correlated qPCR data from 10 patients with tumor sample matched snap-frozen ovarian carcinoma samples with the microarray data from TCGA. We normalized results to the average expression of three housekeeping genes. We scaled and centered the data for comparison to the Affymetrix output.
For the 18 specimens, gene expression data obtained from snap-frozen tissue correlated highly with that from FFPE samples in our TaqMan assay (r > 0.82). For the 10 duplicate TCGA specimens, the reported microarray data correlated well (r = 0.6) with our qPCR data, and ranges of expression along pathways were similar.
Gene expression data obtained by qPCR from FFPE serous ovarian carcinoma samples can be used to assess in the pathway-based predictive model. The normalization procedures described control variations in expression, and the range calculated along a specific pathway can be interpreted for a patient's risk profile.
此前,我们利用来自癌症基因组图谱(TCGA)的临床和基因表达数据构建了一个基于通路的指标,用于预测卵巢癌的预后。这些数据是通过Affymetrix U133平台对速冻组织进行测量获得的。在本研究中,我们将用于建模的数据与来自TaqMan定量聚合酶链反应(qPCR)的速冻和石蜡包埋(FFPE)样本数据进行了关联。
为了比较保存方法对qPCR测量基因表达的影响,我们评估了18对患者及其肿瘤样本匹配的速冻和FFPE卵巢癌样本。为了比较基因测量技术,我们将10例肿瘤样本匹配的速冻卵巢癌样本的qPCR数据与TCGA的微阵列数据进行了关联。我们将结果标准化为三个管家基因的平均表达量。我们对数据进行了缩放和中心化处理,以便与Affymetrix的输出结果进行比较。
对于这18个样本,在我们的TaqMan检测中,从速冻组织获得的基因表达数据与FFPE样本的基因表达数据高度相关(r>0.82)。对于10个TCGA重复样本,报告的微阵列数据与我们的qPCR数据相关性良好(r = 0.6),并且沿通路的表达范围相似。
通过qPCR从FFPE浆液性卵巢癌样本中获得的基因表达数据可用于基于通路的预测模型评估。所描述的标准化程序可控制表达差异,并且沿特定通路计算的范围可用于解读患者的风险概况。