Schachtschneider Kyle M, Madsen Ole, Park Chankyu, Rund Laurie A, Groenen Martien A M, Schook Lawrence B
Department of Animal Sciences, University of Illinois, Urbana, IL, USA.
Animal Breeding and Genomics Center, Wageningen University, Wageningen, The Netherlands.
BMC Genomics. 2015 Oct 5;16:743. doi: 10.1186/s12864-015-1938-x.
Pigs (Sus scrofa) provide relevant biomedical models to dissect complex diseases due to their anatomical, genetic, and physiological similarities with humans. Aberrant DNA methylation has been linked to many of these diseases and is associated with gene expression; however, the functional similarities and differences between porcine and human DNA methylation patterns are largely unknown.
DNA and RNA was isolated from eight tissue samples (fat, heart, kidney, liver, lung, lymph node, muscle, and spleen) from the adult female Duroc utilized for the pig genome sequencing project. Reduced representation bisulfite sequencing (RRBS) and RNA-seq were performed on an Illumina HiSeq2000. RRBS reads were aligned using BSseeker2, and only sites with a minimum depth of 10 reads were used for methylation analysis. RNA-seq reads were aligned using Tophat, and expression analysis was performed using Cufflinks. In addition, SNP calling was performed using GATK for targeted control and whole genome sequencing reads for CpG site validation and allelic expression analysis, respectively.
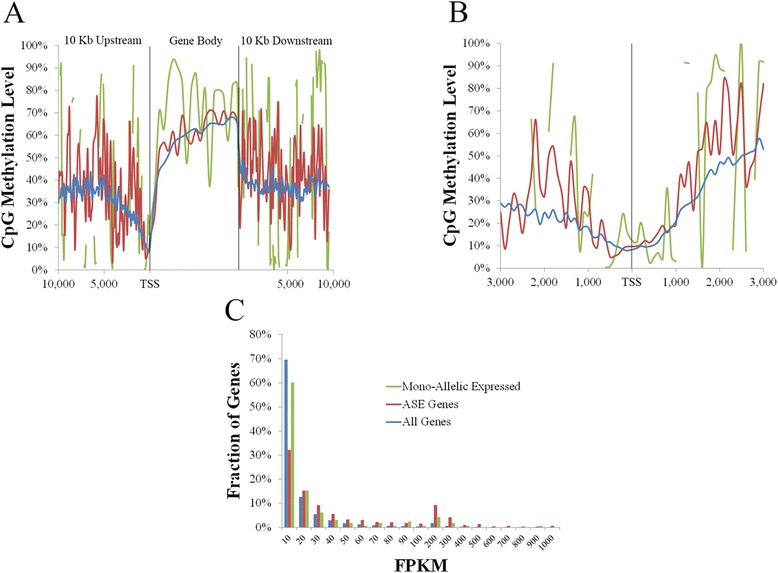
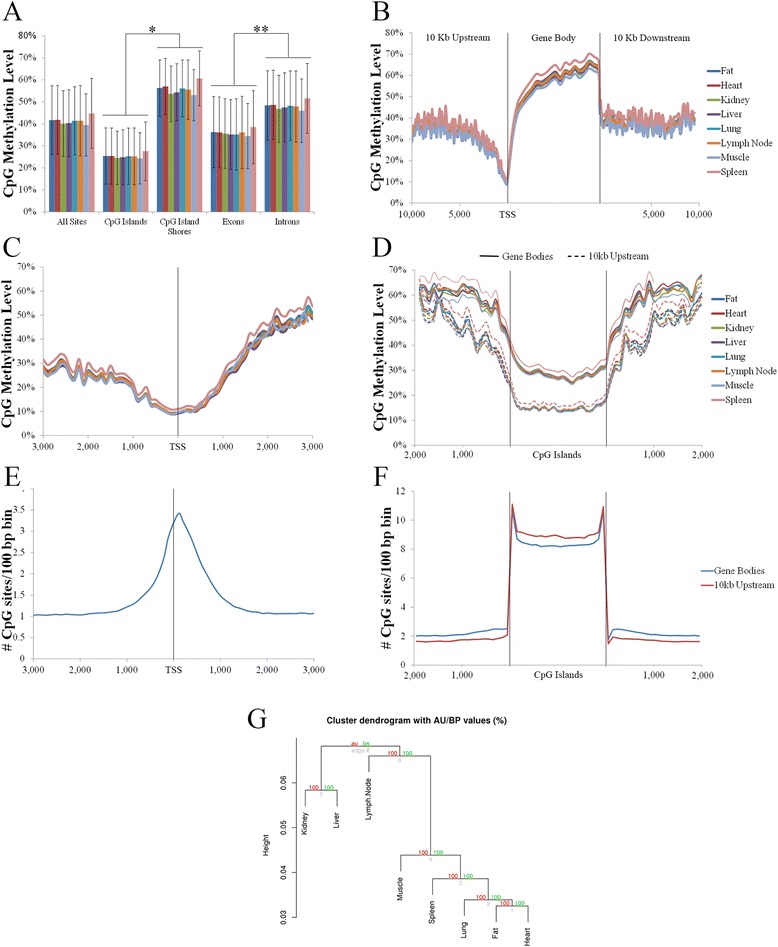
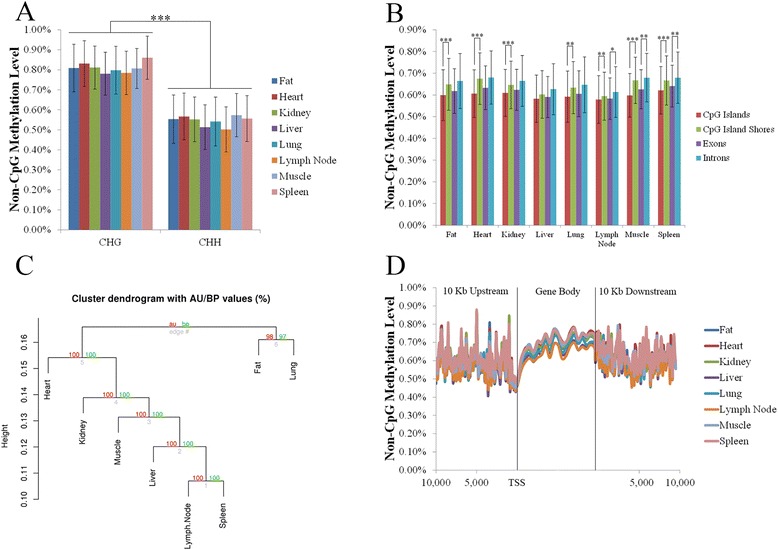
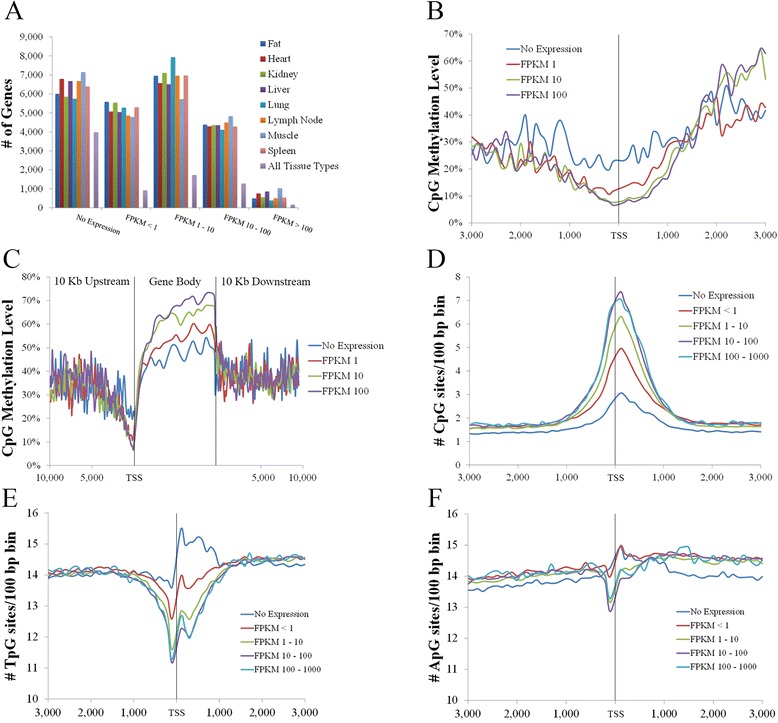
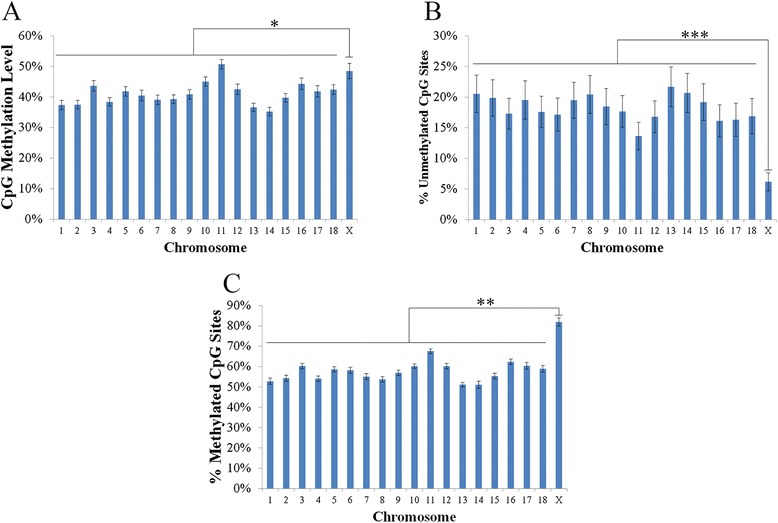
Analysis on the influence of DNA variation in methylation calling revealed a reduced effectiveness of WGS datasets in covering CpG rich regions, as well as the usefulness of a targeted control library for SNP detection. Analysis of over 500,000 CpG sites demonstrated genome wide methylation patterns similar to those observed in humans, including reduced methylation within CpG islands and at transcription start sites (TSS), X chromosome inactivation, and anticorrelation of TSS CpG methylation with gene expression. In addition, a positive correlation between TSS CpG density and expression, and a negative correlation between TSS TpG density and expression were demonstrated. Low but non-random non-CpG methylation (<1%) was also detected in all non-neuronal somatic tissues, with differences in tissue clustering observed based on CpG and non-CpG methylation patterns. Finally, allele specific expression analysis revealed enrichment of genes involved in metabolic and regulatory processes.
These results provide transcriptional and DNA methylation datasets for the biomedical community that are directly relatable to current genomic resources. In addition, the correlation between TSS CpG density and expression suggests increased mutation rates at CpG sites play a significant role in adaptive evolution by reducing CpG density at TSS over time, resulting in higher methylation levels in these regions and more permanent changes to lower gene expression. This is proposed to occur predominantly through deamination of 5-methylcytosine to thymidine, resulting in the replacement of CpG with TpG sites in these regions, as indicated by the increased TSS TpG density observed in non-expressed genes, resulting in a negative correlation between expression and TSS TpG density.
This study provides baseline methylation and gene transcription profiles for a healthy adult pig, reports similar patterns to those observed in humans, and supports future porcine studies related to human disease and development. Additionally, the observed reduced CpG and increased TpG density at TSS of lowly expressed genes suggests DNA methylation plays a significant role in adaptive evolution through more permanent changes to lower gene expression.
猪(野猪)由于在解剖学、遗传学和生理学上与人类相似,为剖析复杂疾病提供了相关的生物医学模型。异常的DNA甲基化与许多此类疾病相关,并与基因表达有关;然而,猪和人类DNA甲基化模式之间的功能异同很大程度上尚不清楚。
从用于猪基因组测序项目的成年雌性杜洛克猪的八个组织样本(脂肪、心脏、肾脏、肝脏、肺、淋巴结、肌肉和脾脏)中分离DNA和RNA。在Illumina HiSeq2000上进行简化代表性亚硫酸氢盐测序(RRBS)和RNA测序。使用BSseeker2对RRBS读数进行比对,仅将最低深度为10次读数的位点用于甲基化分析。使用Tophat对RNA测序读数进行比对,并使用Cufflinks进行表达分析。此外,分别使用GATK进行单核苷酸多态性(SNP)检测以用于靶向对照,并使用全基因组测序读数进行CpG位点验证和等位基因表达分析。
对DNA变异在甲基化检测中的影响分析表明,全基因组测序数据集在覆盖富含CpG的区域方面有效性降低,以及靶向对照文库在SNP检测中的有用性。对超过500,000个CpG位点的分析表明,全基因组甲基化模式与在人类中观察到的相似,包括CpG岛和转录起始位点(TSS)内甲基化减少、X染色体失活以及TSS CpG甲基化与基因表达的反相关。此外,还证明了TSS CpG密度与表达之间呈正相关,以及TSS TpG密度与表达之间呈负相关。在所有非神经体细胞组织中也检测到低但非随机的非CpG甲基化(<1%),基于CpG和非CpG甲基化模式观察到组织聚类存在差异。最后,等位基因特异性表达分析揭示了参与代谢和调节过程的基因富集。
这些结果为生物医学领域提供了与当前基因组资源直接相关的转录和DNA甲基化数据集。此外,TSS CpG密度与表达之间的相关性表明,随着时间的推移,CpG位点的突变率增加通过降低TSS处的CpG密度在适应性进化中起重要作用,导致这些区域甲基化水平更高以及基因表达发生更持久的变化。这被认为主要通过5-甲基胞嘧啶脱氨生成胸腺嘧啶而发生,导致这些区域中CpG被TpG位点取代,如在未表达基因中观察到的TSS TpG密度增加所示,从而导致表达与TSS TpG密度之间呈负相关。
本研究提供了健康成年猪的基线甲基化和基因转录谱,报告了与人类中观察到的相似模式,并支持未来与人类疾病和发育相关的猪研究。此外,在低表达基因的TSS处观察到的CpG减少和TpG密度增加表明DNA甲基化通过对基因表达进行更持久的改变在适应性进化中起重要作用。