Feldbauer Roman, Schulz Frederik, Horn Matthias, Rattei Thomas
BMC Bioinformatics. 2015;16 Suppl 14(Suppl 14):S1. doi: 10.1186/1471-2105-16-S14-S1. Epub 2015 Oct 2.
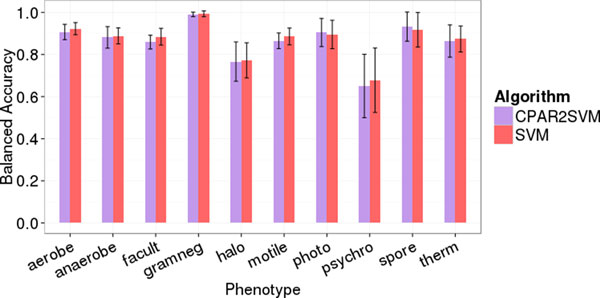
The accessibility of almost complete genome sequences of uncultivable microbial species from metagenomes necessitates computational methods predicting microbial phenotypes solely based on genomic data. Here we investigate how comparative genomics can be utilized for the prediction of microbial phenotypes. The PICA framework facilitates application and comparison of different machine learning techniques for phenotypic trait prediction. We have improved and extended PICA's support vector machine plug-in and suggest its applicability to large-scale genome databases and incomplete genome sequences. We have demonstrated the stability of the predictive power for phenotypic traits, not perturbed by the rapid growth of genome databases. A new software tool facilitates the in-depth analysis of phenotype models, which associate expected and unexpected protein functions with particular traits. Most of the traits can be reliably predicted in only 60-70% complete genomes. We have established a new phenotypic model that predicts intracellular microorganisms. Thereby we could demonstrate that also independently evolved phenotypic traits, characterized by genome reduction, can be reliably predicted based on comparative genomics. Our results suggest that the extended PICA framework can be used to automatically annotate phenotypes in near-complete microbial genome sequences, as generated in large numbers in current metagenomics studies.
来自宏基因组的不可培养微生物物种几乎完整的基因组序列的可获取性,使得仅基于基因组数据预测微生物表型的计算方法成为必要。在此,我们研究如何利用比较基因组学来预测微生物表型。PICA框架有助于应用和比较用于表型特征预测的不同机器学习技术。我们改进并扩展了PICA的支持向量机插件,并表明其适用于大规模基因组数据库和不完整基因组序列。我们已经证明了表型特征预测能力的稳定性,不受基因组数据库快速增长的干扰。一个新的软件工具便于对表型模型进行深入分析,该模型将预期和意外的蛋白质功能与特定特征联系起来。大多数特征在基因组仅60 - 70%完整时就能可靠预测。我们建立了一个预测细胞内微生物的新表型模型。由此我们可以证明,基于比较基因组学,以基因组缩减为特征的独立进化的表型特征也能可靠预测。我们的结果表明,扩展的PICA框架可用于自动注释当前宏基因组学研究中大量生成的近乎完整的微生物基因组序列中的表型。