Thome Aaron D, Standaert David G, Harms Ashley S
Center for Neurodegeneration and Experimental Therapeutics, Department of Neurology, The University of Alabama at Birmingham, Birmingham, Alabama, United States of America.
PLoS One. 2015 Oct 15;10(10):e0140566. doi: 10.1371/journal.pone.0140566. eCollection 2015.
Parkinson disease (PD) is a progressive neurodegenerative disorder characterized by loss of dopamine neurons in the substantia nigra pars compacta (SNpc) and widespread aggregates of the protein alpha-synuclein (α-syn). Increasing evidence points to inflammation as a chief mediator; however, the role of α-syn in triggering and sustaining inflammation remains unclear. In models of Alzheimer's disease (AD), multiple sclerosis (MS) and neurotoxin models of PD, the chemokine CX3CL1 (fractalkine) and its receptor (CX3CR1) have important roles in modulating neuroinflammation.
To examine the role of fractalkine signaling in α-syn-induced-neuroinflammation and neurodegeneration, we used an in vivo mouse model in which human α-syn is overexpressed by an adeno associated viral vector serotype 2 (AAV2) and in vitro phagocytosis and protein internalization assays with primary microglia treated with aggregated α-syn.
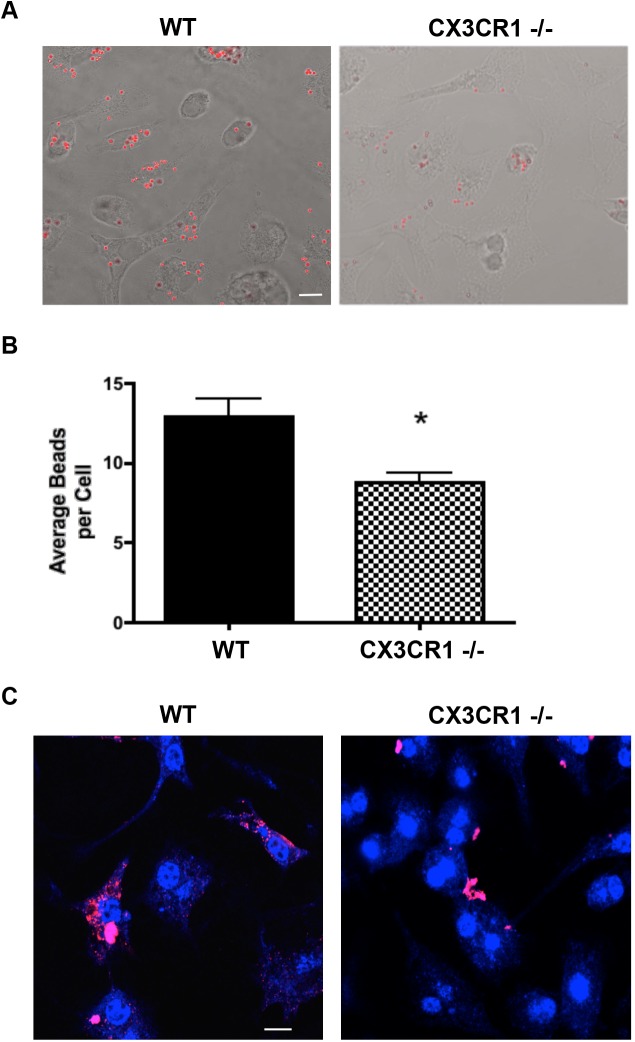
We observed that loss of CX3CR1 expression led to a reduced inflammatory response, with reduced IgG deposition and expression of MHCII 4 weeks post-transduction. Six months post transduction, AAV2 mediated overexpression of α-syn leads to loss of dopaminergic neurons, and this loss was not exacerbated in animals with deletion of CX3CR1. To determine the mechanism by which CX3CR1affects inflammatory responses in α-syn-induced inflammation, phagocytosis was assessed using a fluorescent microsphere assay as well as by microglial uptake of aggregated α-syn. CX3CR1-/- microglia showed reduced uptake of fluorescent beads and aggregated α-syn.
Our results suggest that one mechanism by which CX3CR1-/- attenuates inflammation is at the level of phagocytosis of aggregated α-syn by microglia. These data implicate fractalkine signaling as a potential therapeutic target for regulating inflammatory response in α-syn models PD.
帕金森病(PD)是一种进行性神经退行性疾病,其特征是黑质致密部(SNpc)中的多巴胺能神经元丧失以及蛋白质α-突触核蛋白(α-syn)广泛聚集。越来越多的证据表明炎症是主要介质;然而,α-syn在引发和维持炎症中的作用仍不清楚。在阿尔茨海默病(AD)、多发性硬化症(MS)模型以及PD的神经毒素模型中,趋化因子CX3CL1(分形素)及其受体(CX3CR1)在调节神经炎症中发挥重要作用。
为了研究分形素信号在α-syn诱导的神经炎症和神经退行性变中的作用,我们使用了一种体内小鼠模型,其中人α-syn由2型腺相关病毒载体(AAV2)过表达,并对用聚集的α-syn处理的原代小胶质细胞进行体外吞噬和蛋白质内化试验。
我们观察到,CX3CR1表达缺失导致炎症反应减弱,转导后4周IgG沉积和MHCII表达减少。转导后6个月,AAV2介导的α-syn过表达导致多巴胺能神经元丧失,而在CX3CR1缺失的动物中这种丧失并未加剧。为了确定CX3CR1影响α-syn诱导炎症中炎症反应的机制,使用荧光微球试验以及小胶质细胞对聚集的α-syn的摄取来评估吞噬作用。CX3CR1基因敲除的小胶质细胞显示出对荧光珠和聚集的α-syn的摄取减少。
我们的结果表明,CX3CR1基因敲除减轻炎症的一种机制是在小胶质细胞对聚集α-syn的吞噬水平。这些数据表明分形素信号作为调节α-syn模型PD中炎症反应的潜在治疗靶点。