Ross-Adams H, Lamb A D, Dunning M J, Halim S, Lindberg J, Massie C M, Egevad L A, Russell R, Ramos-Montoya A, Vowler S L, Sharma N L, Kay J, Whitaker H, Clark J, Hurst R, Gnanapragasam V J, Shah N C, Warren A Y, Cooper C S, Lynch A G, Stark R, Mills I G, Grönberg H, Neal D E
Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge CB2 0RE, UK.
Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge CB2 0RE, UK ; Department of Urology, Addenbrooke's Hospital, Cambridge CB2 2QQ, UK ; Academic Urology Group, University of Cambridge, Cambridge, CB2 0QQ, UK.
EBioMedicine. 2015 Jul 29;2(9):1133-44. doi: 10.1016/j.ebiom.2015.07.017. eCollection 2015 Sep.
Understanding the heterogeneous genotypes and phenotypes of prostate cancer is fundamental to improving the way we treat this disease. As yet, there are no validated descriptions of prostate cancer subgroups derived from integrated genomics linked with clinical outcome.
In a study of 482 tumour, benign and germline samples from 259 men with primary prostate cancer, we used integrative analysis of copy number alterations (CNA) and array transcriptomics to identify genomic loci that affect expression levels of mRNA in an expression quantitative trait loci (eQTL) approach, to stratify patients into subgroups that we then associated with future clinical behaviour, and compared with either CNA or transcriptomics alone.
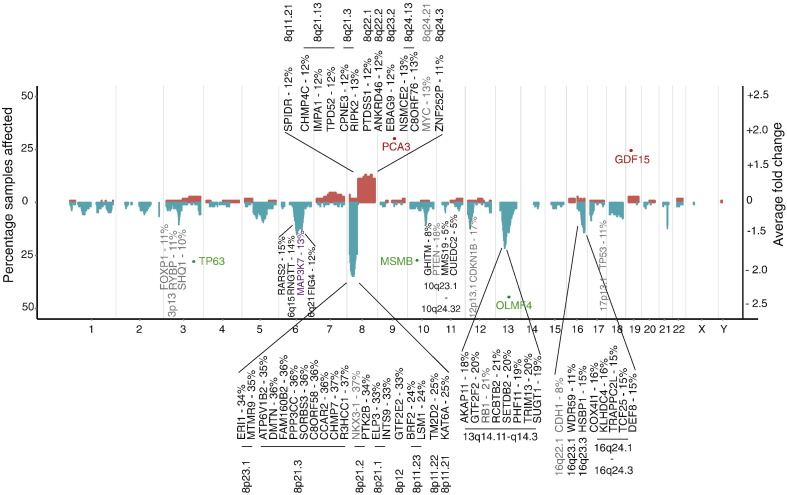
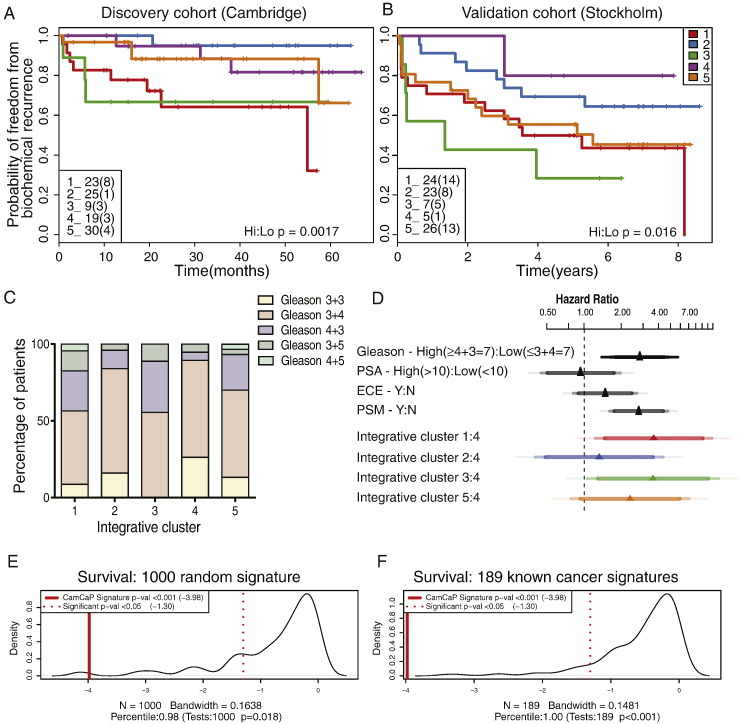
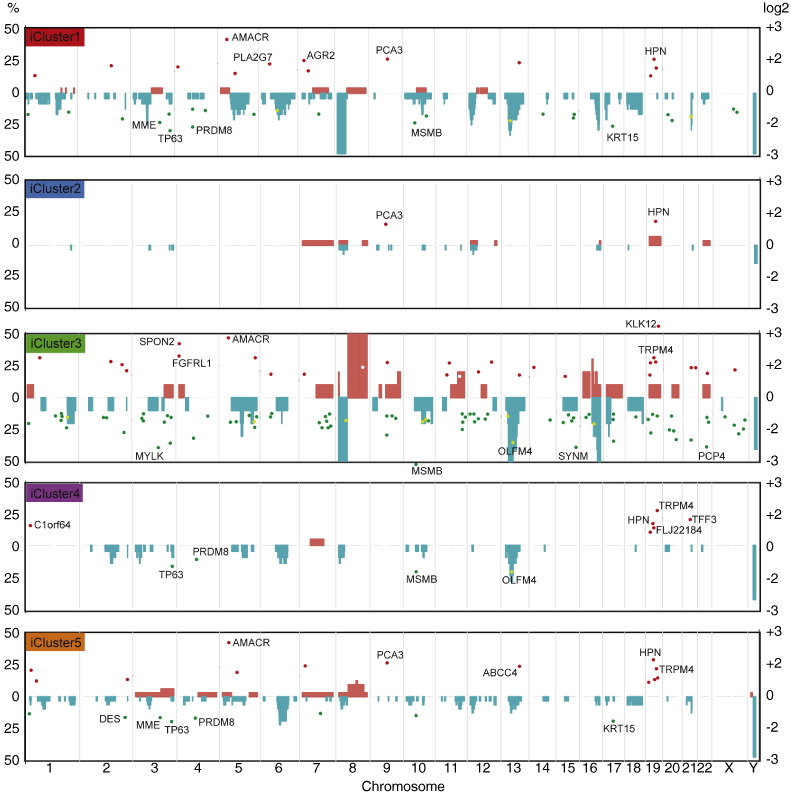
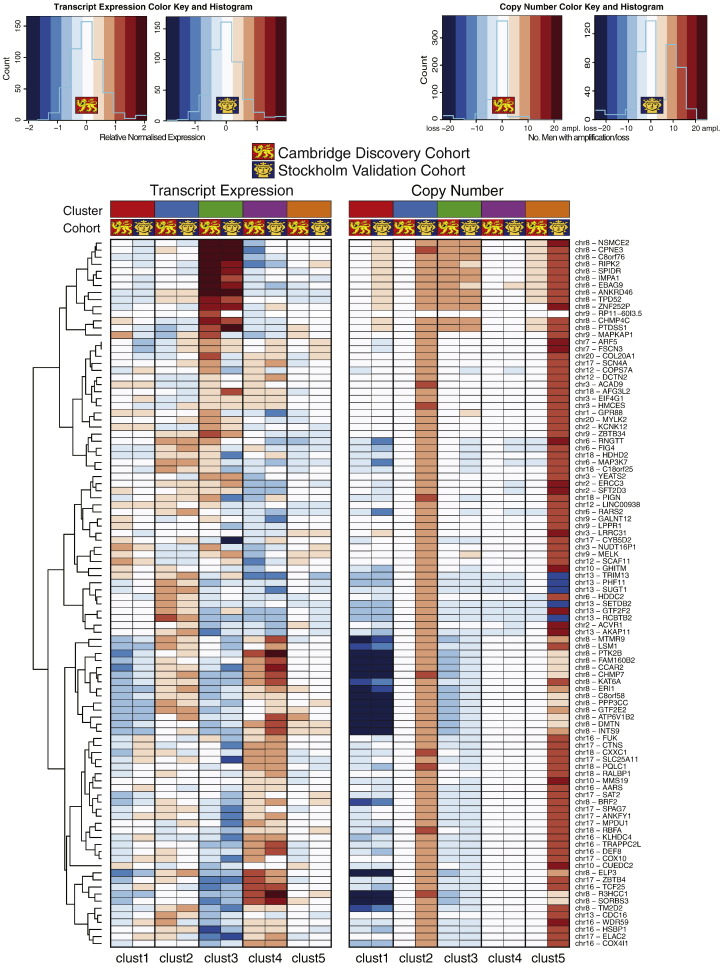
We identified five separate patient subgroups with distinct genomic alterations and expression profiles based on 100 discriminating genes in our separate discovery and validation sets of 125 and 103 men. These subgroups were able to consistently predict biochemical relapse (p = 0.0017 and p = 0.016 respectively) and were further validated in a third cohort with long-term follow-up (p = 0.027). We show the relative contributions of gene expression and copy number data on phenotype, and demonstrate the improved power gained from integrative analyses. We confirm alterations in six genes previously associated with prostate cancer (MAP3K7, MELK, RCBTB2, ELAC2, TPD52, ZBTB4), and also identify 94 genes not previously linked to prostate cancer progression that would not have been detected using either transcript or copy number data alone. We confirm a number of previously published molecular changes associated with high risk disease, including MYC amplification, and NKX3-1, RB1 and PTEN deletions, as well as over-expression of PCA3 and AMACR, and loss of MSMB in tumour tissue. A subset of the 100 genes outperforms established clinical predictors of poor prognosis (PSA, Gleason score), as well as previously published gene signatures (p = 0.0001). We further show how our molecular profiles can be used for the early detection of aggressive cases in a clinical setting, and inform treatment decisions.
For the first time in prostate cancer this study demonstrates the importance of integrated genomic analyses incorporating both benign and tumour tissue data in identifying molecular alterations leading to the generation of robust gene sets that are predictive of clinical outcome in independent patient cohorts.
了解前列腺癌的异质基因型和表型是改善我们治疗这种疾病方式的基础。目前,尚无来自与临床结果相关的整合基因组学的前列腺癌亚组的有效描述。
在一项对259名原发性前列腺癌男性的482份肿瘤、良性和种系样本的研究中,我们使用拷贝数改变(CNA)和阵列转录组学的综合分析,以表达定量性状位点(eQTL)方法识别影响mRNA表达水平的基因组位点,将患者分层为亚组,然后将这些亚组与未来的临床行为相关联,并与单独的CNA或转录组学进行比较。
在我们分别为125名和103名男性的发现和验证集中,基于100个区分基因,我们确定了五个具有不同基因组改变和表达谱的独立患者亚组。这些亚组能够一致地预测生化复发(分别为p = 0.0017和p = 0.016),并在第三个进行长期随访的队列中得到进一步验证(p = 0.027)。我们展示了基因表达和拷贝数数据对表型的相对贡献,并证明了综合分析所获得的更强效力。我们证实了先前与前列腺癌相关的六个基因(MAP3K7、MELK、RCBTB2、ELAC2、TPD52、ZBTB4)的改变,还鉴定了94个先前未与前列腺癌进展相关的基因,这些基因仅使用转录本或拷贝数数据是无法检测到的。我们证实了一些先前发表过的与高危疾病相关的分子变化,包括MYC扩增、NKX3-1、RB1和PTEN缺失,以及肿瘤组织中PCA3和AMACR的过表达和MSMB的缺失。这100个基因中的一个子集优于已确立的预后不良临床预测指标(PSA、Gleason评分)以及先前发表的基因特征(p = 0.0001)。我们进一步展示了我们的分子谱如何用于临床环境中侵袭性病例的早期检测,并为治疗决策提供依据。
本研究在前列腺癌中首次证明了整合基因组分析的重要性,该分析纳入良性和肿瘤组织数据,以识别导致生成强大基因集的分子改变,这些基因集可预测独立患者队列中的临床结果。