Cao Renzhi, Cheng Jianlin
Computer Science Department, University of Missouri, Columbia, Missouri, 65211, USA.
Informatics Institute, University of Missouri, Columbia, Missouri, 65211, USA.
BMC Genomics. 2015 Oct 28;16:880. doi: 10.1186/s12864-015-2093-0.
A number of factors have been investigated in the context of gene function prediction and analysis, such as sequence identity, gene expressions, and gene co-evolution. However, three-dimensional (3D) conformation of the genome has not been tapped to analyse gene function, probably largely due to lack of genome conformation data until recently.
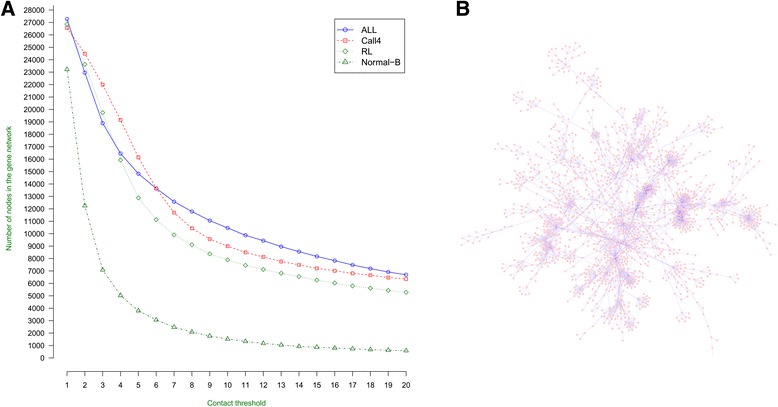
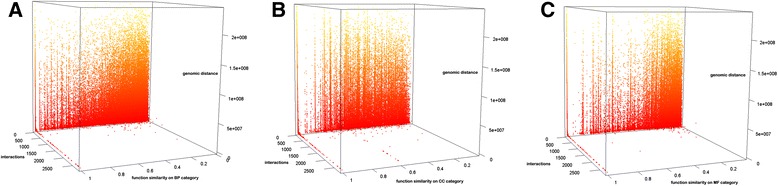
We construct the genome-wide spatial gene-gene interaction networks for three different human B-cells or cell lines from their chromosomal contact data generated by the Hi-C chromosome conformation capturing technique. The G-SESAME and Fast-SemSim are used to calculate function similarity between interacted / non-interacted genes. The Gene Ontology statistics computed from the gene-gene interaction networks is used for gene function prediction.
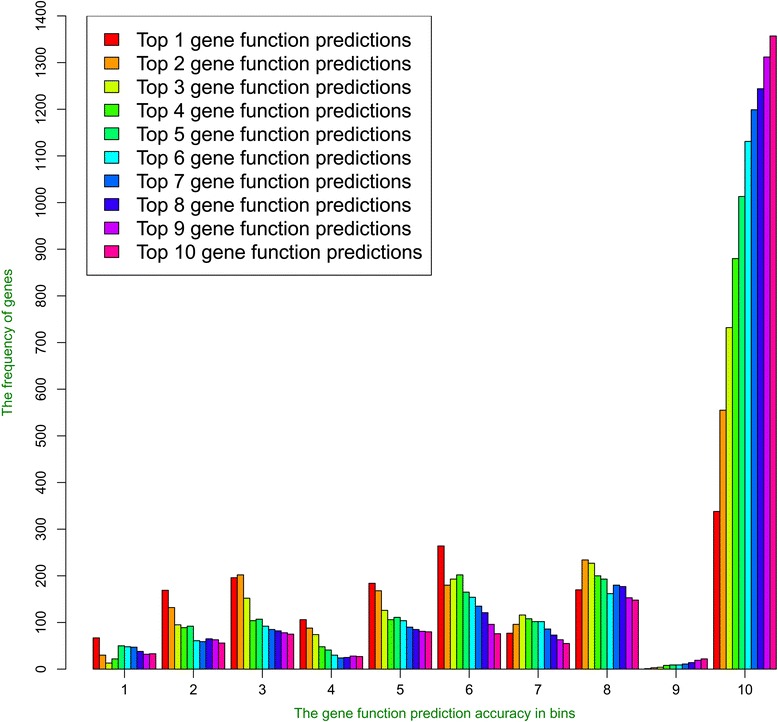
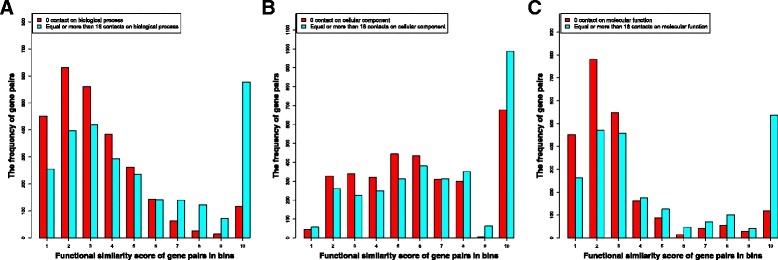
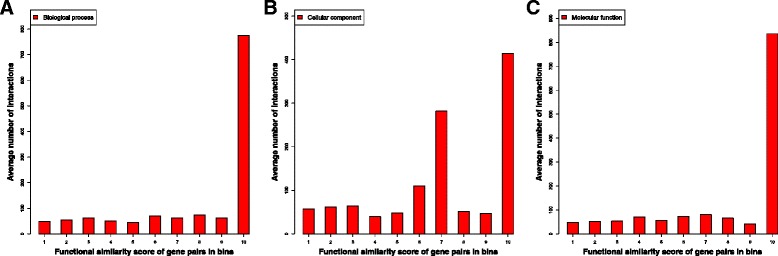
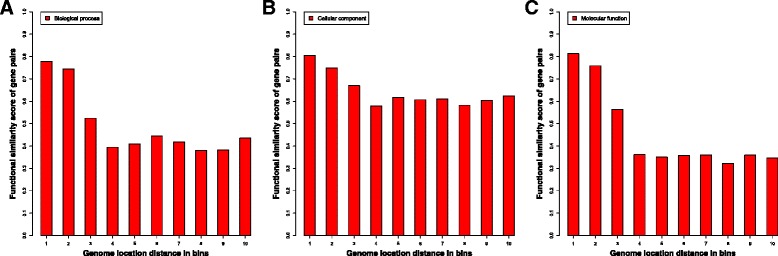
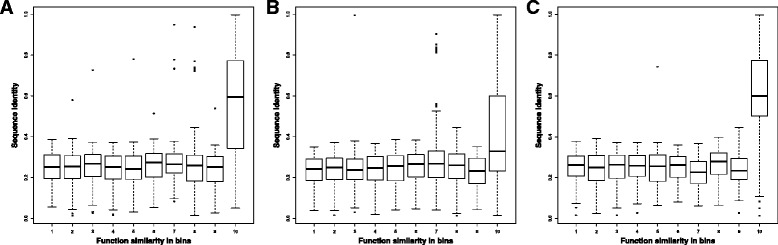
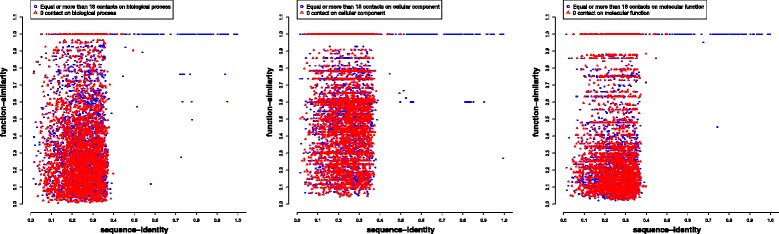
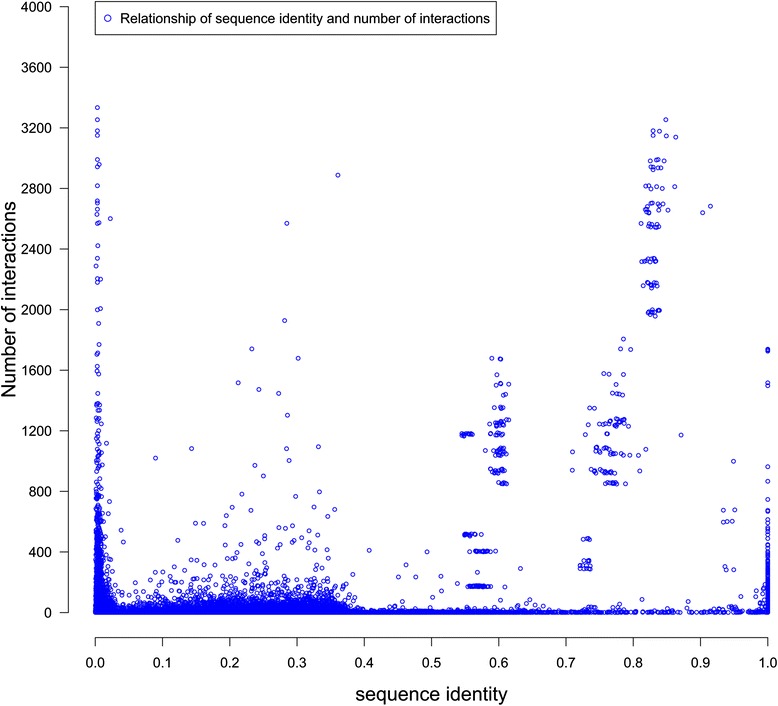
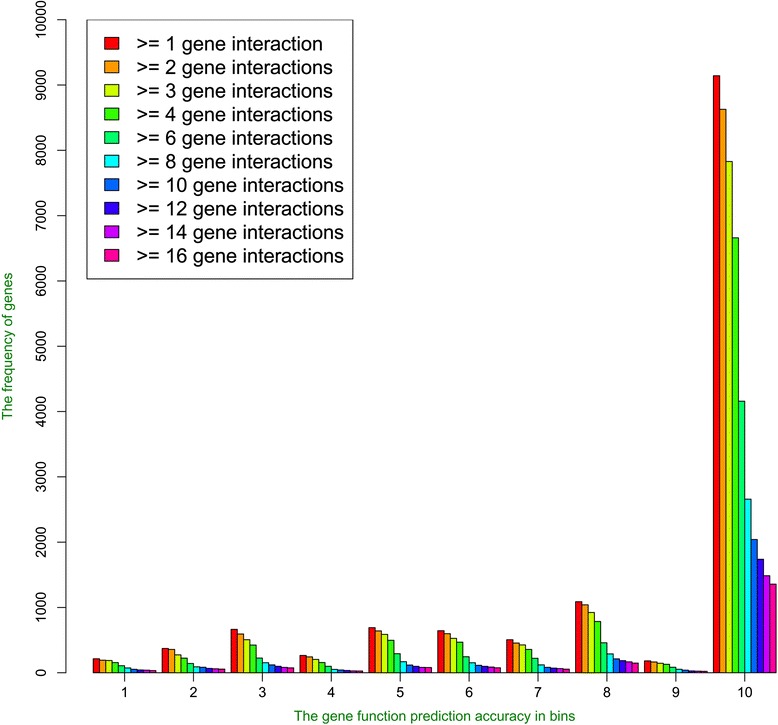
We compare the function similarity of gene pairs that do not spatially interact and that have interactions. We find that genes that have strong spatial interactions tend to have highly similar function in terms of biological process, molecular function and cellular component of the Gene Ontology. And even though the level of gene-gene interactions generally have no or weak correlation with either sequential genomic distance or sequence identity between genes, the interacted genes with high function similarity tend to have stronger interactions, somewhat shorter genomic distance and significantly higher sequence identity. And combining genomic distance or sequence identity with spatial gene-gene interaction information informs gene-gene function similarity much better than using either one of them alone, suggesting gene-gene interaction information is largely complementary with genomic distance and sequence identity in the context of gene function analysis. We develop and evaluate a new gene function prediction method based on gene-gene interacting networks, which can predict gene function well for a large number of human genes.
In this work, we demonstrate that the spatial conformation of the human genome is relevant to gene function similarity and is useful for gene function prediction.
在基因功能预测和分析的背景下,人们已经研究了许多因素,如序列同一性、基因表达和基因共同进化。然而,基因组的三维(3D)构象尚未被用于分析基因功能,这可能主要是因为直到最近才缺乏基因组构象数据。
我们利用Hi-C染色体构象捕获技术生成的染色体接触数据,构建了三种不同人类B细胞或细胞系的全基因组空间基因-基因相互作用网络。使用G-SESAME和Fast-SemSim计算相互作用/非相互作用基因之间的功能相似性。从基因-基因相互作用网络计算得到的基因本体统计数据用于基因功能预测。
我们比较了在空间上不相互作用和有相互作用的基因对的功能相似性。我们发现,在基因本体的生物学过程、分子功能和细胞成分方面,具有强空间相互作用的基因往往具有高度相似的功能。而且,尽管基因-基因相互作用水平通常与基因间的序列基因组距离或序列同一性没有或只有微弱的相关性,但具有高功能相似性的相互作用基因往往具有更强的相互作用、稍短的基因组距离和显著更高的序列同一性。将基因组距离或序列同一性与空间基因-基因相互作用信息相结合,比单独使用其中任何一个能更好地反映基因-基因功能相似性,这表明在基因功能分析中,基因-基因相互作用信息在很大程度上与基因组距离和序列同一性是互补的。我们开发并评估了一种基于基因-基因相互作用网络的新基因功能预测方法,该方法可以很好地预测大量人类基因的功能。
在这项工作中,我们证明了人类基因组的空间构象与基因功能相似性相关,并且对基因功能预测有用。