Bertoli Sarah, Boutzen Helena, David Laure, Larrue Clément, Vergez François, Fernandez-Vidal Anne, Yuan Lingli, Hospital Marie-Anne, Tamburini Jérôme, Demur Cécile, Delabesse Eric, Saland Estelle, Sarry Jean-Emmanuel, Galcera Marie-Odile, Mansat-De Mas Véronique, Didier Christine, Dozier Christine, Récher Christian, Manenti Stéphane
Cancer Research Center of Toulouse, Inserm UMR 1037, CNRS ERL 5294, Université de Toulouse, Oncopole, Toulouse, France.
Hematology Department, Institut Universitaire du Cancer Toulouse - Oncopole, Toulouse, France.
Oncotarget. 2015 Nov 10;6(35):38061-78. doi: 10.18632/oncotarget.5706.
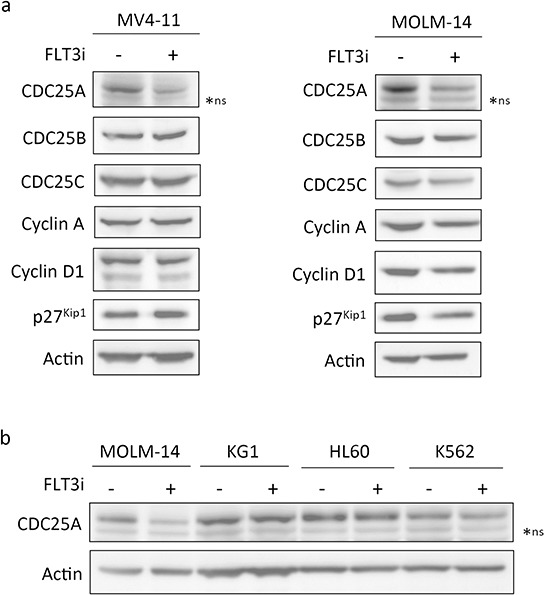
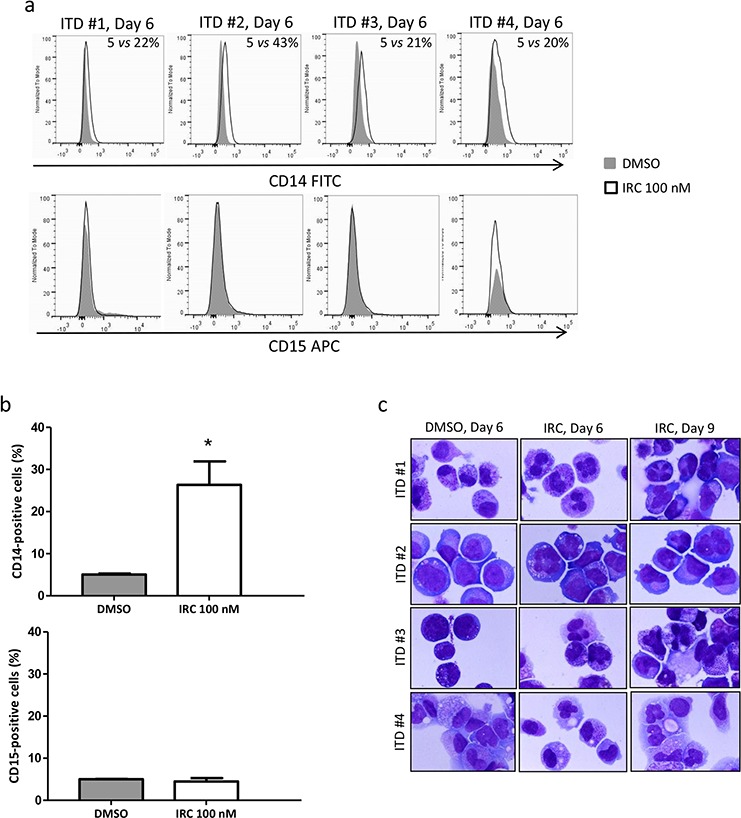
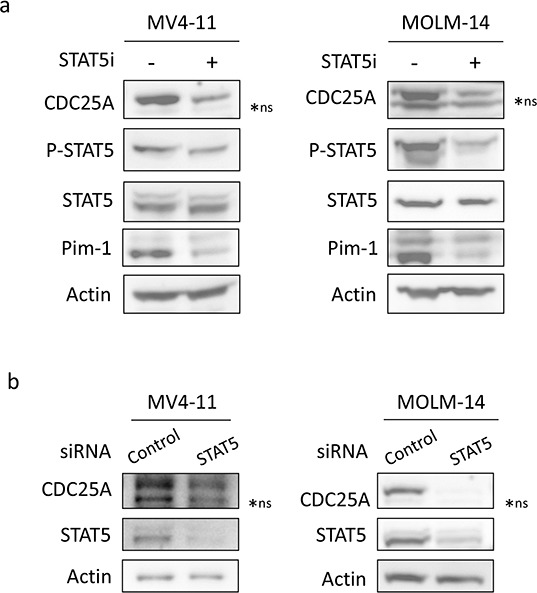
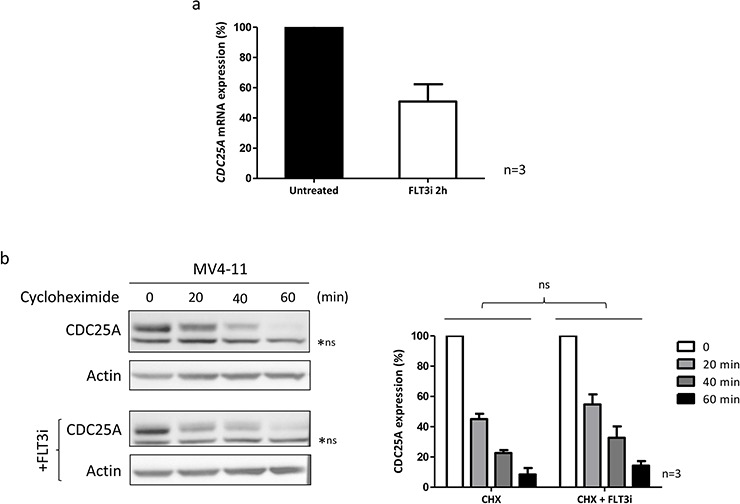
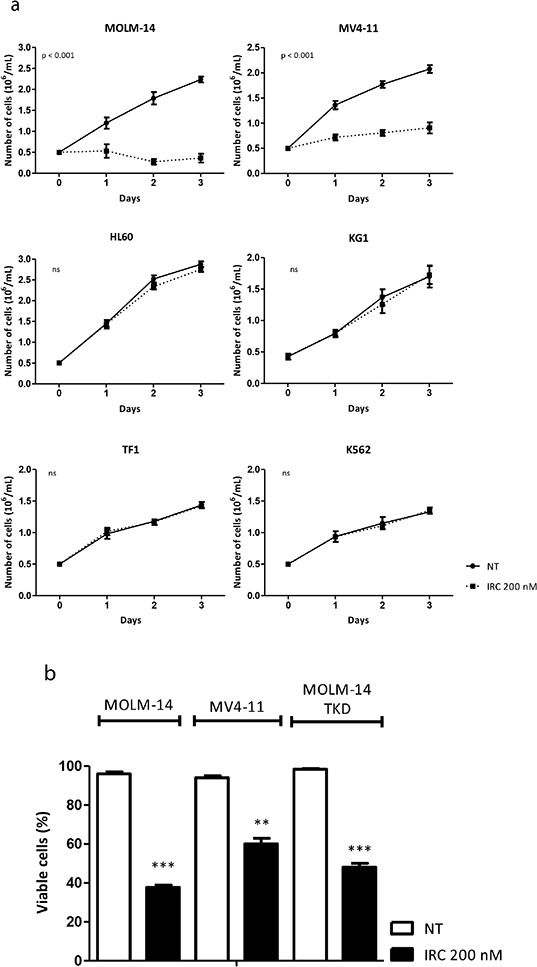
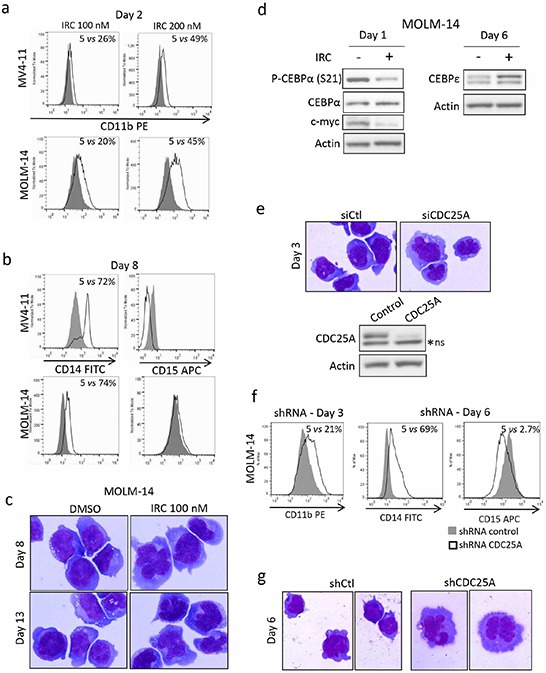
We investigated cell cycle regulation in acute myeloid leukemia cells expressing the FLT3-ITD mutated tyrosine kinase receptor, an underexplored field in this disease. Upon FLT3 inhibition, CDC25A mRNA and protein were rapidly down-regulated, while levels of other cell cycle proteins remained unchanged. This regulation was dependent on STAT5, arguing for FLT3-ITD-dependent transcriptional regulation of CDC25A. CDC25 inhibitors triggered proliferation arrest and cell death of FLT3-ITD as well as FLT3-ITD/TKD AC-220 resistant cells, but not of FLT3-wt cells. Consistently, RNA interference-mediated knock-down of CDC25A reduced the proliferation of FLT3-ITD cell lines. Finally, the clonogenic capacity of primary FLT3-ITD AML cells was reduced by the CDC25 inhibitor IRC-083864, while FLT3-wt AML and normal CD34+ myeloid cells were unaffected. In good agreement, in a cohort of 100 samples from AML patients with intermediate-risk cytogenetics, high levels of CDC25A mRNA were predictive of higher clonogenic potential in FLT3-ITD+ samples, not in FLT3-wt ones.Importantly, pharmacological inhibition as well as RNA interference-mediated knock-down of CDC25A also induced monocytic differentiation of FLT3-ITD positive cells, as judged by cell surface markers expression, morphological modifications, and C/EBPα phosphorylation. CDC25 inhibition also re-induced monocytic differentiation in primary AML blasts carrying the FLT3-ITD mutation, but not in blasts expressing wild type FLT3. Altogether, these data identify CDC25A as an early cell cycle transducer of FLT3-ITD oncogenic signaling, and as a promising target to inhibit proliferation and re-induce differentiation of FLT3-ITD AML cells.
我们研究了表达FLT3-ITD突变型酪氨酸激酶受体的急性髓系白血病细胞中的细胞周期调控,这是该疾病中一个尚未充分探索的领域。在FLT3受到抑制后,CDC25A的mRNA和蛋白迅速下调,而其他细胞周期蛋白的水平保持不变。这种调控依赖于STAT5,提示存在FLT3-ITD依赖的CDC25A转录调控。CDC25抑制剂可引发FLT3-ITD以及FLT3-ITD/TKD对AC-220耐药细胞的增殖停滞和细胞死亡,但对FLT3野生型细胞无此作用。同样,RNA干扰介导的CDC25A敲低可降低FLT3-ITD细胞系的增殖。最后,CDC25抑制剂IRC-083864降低了原发性FLT3-ITD AML细胞的克隆形成能力,而FLT3野生型AML细胞和正常CD34+髓系细胞则未受影响。与此一致的是,在100例具有中危细胞遗传学特征的AML患者样本队列中,高水平的CDC25A mRNA可预测FLT3-ITD阳性样本而非FLT3野生型样本具有更高的克隆形成潜力。重要的是,通过细胞表面标志物表达、形态学改变和C/EBPα磷酸化判断,CDC25A的药理学抑制以及RNA干扰介导的敲低还可诱导FLT3-ITD阳性细胞的单核细胞分化。CDC25抑制还可在携带FLT3-ITD突变的原发性AML原始细胞中重新诱导单核细胞分化,但在表达野生型FLT3的原始细胞中则无此作用。总之,这些数据确定CDC25A为FLT3-ITD致癌信号的早期细胞周期转导因子,也是抑制FLT3-ITD AML细胞增殖和重新诱导其分化的一个有前景的靶点。