Wang Mingjie, Doak Thomas G, Ye Yuzhen
School of Informatics and Computing, Indiana University, Bloomington, IN, 47405, USA.
Department of Biology, Indiana University, Bloomington, IN, 47405, USA.
Genome Biol. 2015 Nov 2;16:243. doi: 10.1186/s13059-015-0804-0.
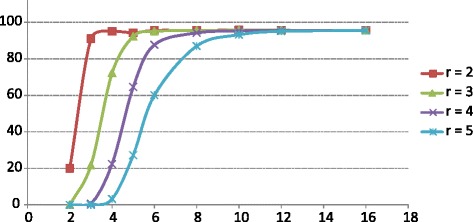
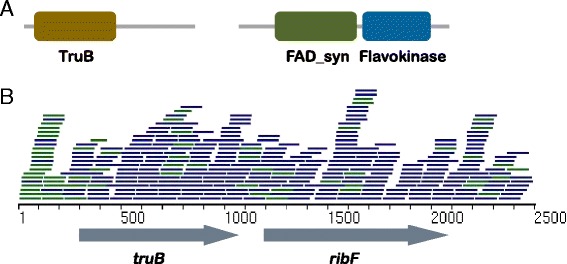
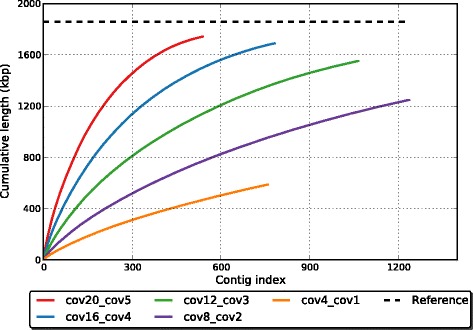
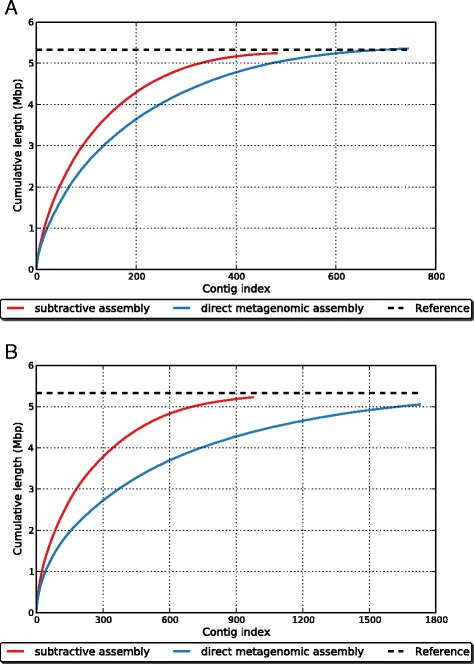
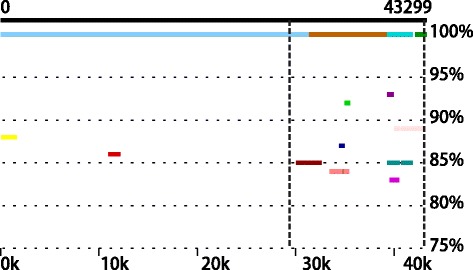
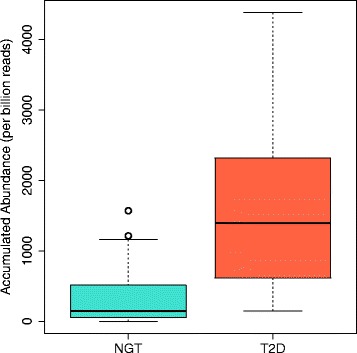
Comparative metagenomics remains challenging due to the size and complexity of metagenomic datasets. Here we introduce subtractive assembly, a de novo assembly approach for comparative metagenomics that directly assembles only the differential reads that distinguish between two groups of metagenomes. Using simulated datasets, we show it improves both the efficiency of the assembly and the assembly quality of the differential genomes and genes. Further, its application to type 2 diabetes (T2D) metagenomic datasets reveals clear signatures of the T2D gut microbiome, revealing new phylogenetic and functional features of the gut microbial communities associated with T2D.
由于宏基因组数据集的规模和复杂性,比较宏基因组学仍然具有挑战性。在这里,我们介绍了减法组装,这是一种用于比较宏基因组学的从头组装方法,它直接仅组装区分两组宏基因组的差异读数。使用模拟数据集,我们表明它提高了组装效率以及差异基因组和基因的组装质量。此外,其在2型糖尿病(T2D)宏基因组数据集上的应用揭示了T2D肠道微生物组的明显特征,揭示了与T2D相关的肠道微生物群落的新系统发育和功能特征。