Konopka Tomasz, Nijman Sebastian M B
Ludwig Institute for Cancer Research, University of Oxford, Oxford, UK.
Bioinformatics. 2016 Mar 1;32(5):657-63. doi: 10.1093/bioinformatics/btv654. Epub 2015 Nov 5.
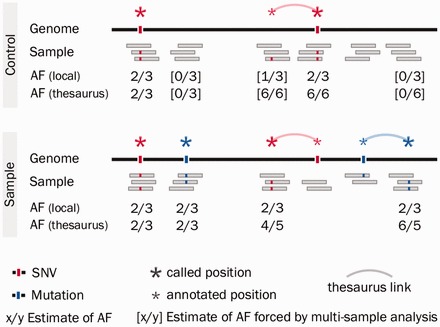
Calling changes in DNA, e.g. as a result of somatic events in cancer, requires analysis of multiple matched sequenced samples. Events in low-mappability regions of the human genome are difficult to encode in variant call files and have been under-reported as a result. However, they can be described accurately through thesaurus annotation-a technique that links multiple genomic loci together to explicate a single variant.
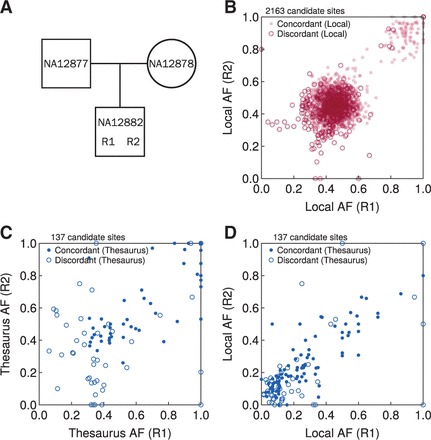
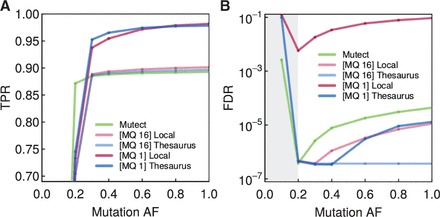
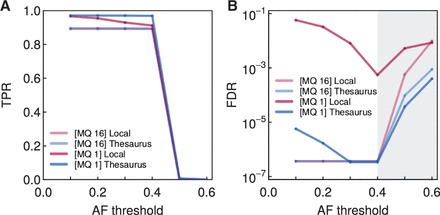
We here describe software and benchmarks for using thesaurus annotation to detect point changes in DNA from matched samples. In benchmarks on matched normal/tumor samples we show that the technique can recover between five and ten percent more true events than conventional approaches, while strictly limiting false discovery and being fully consistent with popular variant analysis workflows. We also demonstrate the utility of the approach for analysis of de novo mutations in parents/child families.
Software performing thesaurus annotation is implemented in java; available in source code on github at GeneticThesaurus (https://github.com/tkonopka/GeneticThesaurus) and as an executable on sourceforge at geneticthesaurus (https://sourceforge.net/projects/geneticthesaurus). Mutation calling is implemented in an R package available on github at RGeneticThesaurus (https://github.com/tkonopka/RGeneticThesaurus).
Supplementary data are available at Bioinformatics online.
识别DNA中的变化,例如癌症中的体细胞事件所导致的变化,需要对多个匹配的测序样本进行分析。人类基因组中低映射性区域的事件难以在变异调用文件中进行编码,因此报告不足。然而,它们可以通过词库注释准确描述——这是一种将多个基因组位点联系在一起以阐明单个变异的技术。
我们在此描述了使用词库注释从匹配样本中检测DNA点变化的软件和基准。在匹配的正常/肿瘤样本基准测试中,我们表明该技术比传统方法能够多发现5%到10%的真实事件,同时严格限制错误发现,并且与流行的变异分析工作流程完全一致。我们还展示了该方法在分析亲子家庭中的新生突变方面的效用。
执行词库注释的软件用Java实现;可在GitHub上的GeneticThesaurus(https://github.com/tkonopka/GeneticThesaurus)获取源代码,也可在SourceForge上的geneticthesaurus(https://sourceforge.net/projects/geneticthesaurus)获取可执行文件。变异调用在GitHub上的RGeneticThesaurus(https://github.com/tkonopka/RGeneticThesaurus)的R包中实现。
补充数据可在《生物信息学》在线获取。