Kerzendorfer Claudia, Konopka Tomasz, Nijman Sebastian M B
Research Center for Molecular Medicine of the Austrian Academy of Sciences (CeMM), Vienna, Austria.
Research Center for Molecular Medicine of the Austrian Academy of Sciences (CeMM), Vienna, Austria
Nucleic Acids Res. 2015 May 26;43(10):e68. doi: 10.1093/nar/gkv178. Epub 2015 Mar 27.
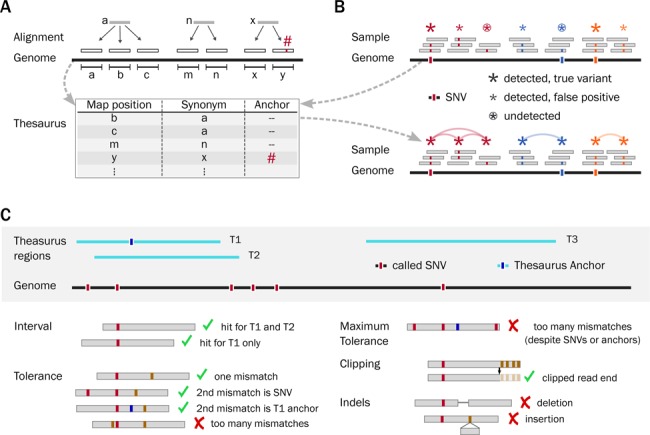
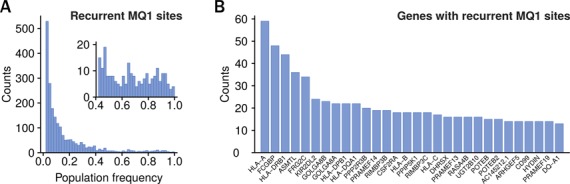
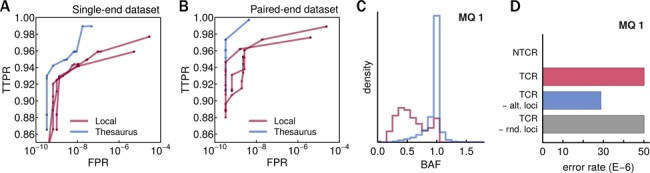
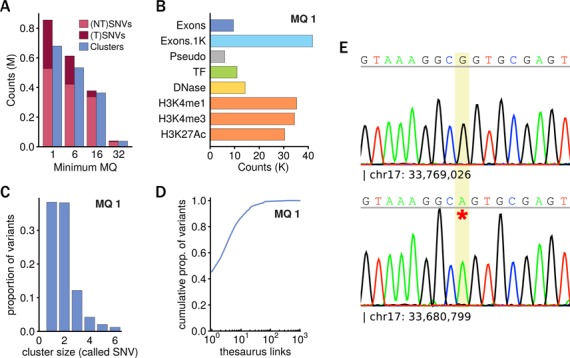
Detecting genetic variation is one of the main applications of high-throughput sequencing, but is still challenging wherever aligning short reads poses ambiguities. Current state-of-the-art variant calling approaches avoid such regions, arguing that it is necessary to sacrifice detection sensitivity to limit false discovery. We developed a method that links candidate variant positions within repetitive genomic regions into clusters. The technique relies on a resource, a thesaurus of genetic variation, that enumerates genomic regions with similar sequence. The resource is computationally intensive to generate, but once compiled can be applied efficiently to annotate and prioritize variants in repetitive regions. We show that thesaurus annotation can reduce the rate of false variant calls due to mappability by up to three orders of magnitude. We apply the technique to whole genome datasets and establish that called variants in low mappability regions annotated using the thesaurus can be experimentally validated. We then extend the analysis to a large panel of exomes to show that the annotation technique opens possibilities to study variation in hereto hidden and under-studied parts of the genome.
检测基因变异是高通量测序的主要应用之一,但在短读长比对存在歧义的任何地方,这仍然具有挑战性。当前最先进的变异检测方法会避开这些区域,认为有必要牺牲检测灵敏度以限制错误发现。我们开发了一种方法,将重复基因组区域内的候选变异位置链接成簇。该技术依赖于一种资源,即基因变异词库,它枚举了具有相似序列的基因组区域。生成该资源计算量很大,但一旦编译完成,就可以有效地应用于注释重复区域中的变异并对其进行优先级排序。我们表明,词库注释可将由于可映射性导致的错误变异调用率降低多达三个数量级。我们将该技术应用于全基因组数据集,并确定使用词库注释的低可映射性区域中调用的变异可以通过实验验证。然后,我们将分析扩展到一大组外显子组,以表明注释技术为研究基因组中迄今隐藏且研究不足的部分的变异开辟了可能性。