Deas Emma, Cremades Nunilo, Angelova Plamena R, Ludtmann Marthe H R, Yao Zhi, Chen Serene, Horrocks Mathew H, Banushi Blerida, Little Daniel, Devine Michael J, Gissen Paul, Klenerman David, Dobson Christopher M, Wood Nicholas W, Gandhi Sonia, Abramov Andrey Y
1 Department of Molecular Neuroscience, UCL Institute of Neurology , Queen Square, London, United Kingdom .
2 Department of Chemistry, Lensfield Road, University of Cambridge , Cambridge, United Kingdom .
Antioxid Redox Signal. 2016 Mar 1;24(7):376-91. doi: 10.1089/ars.2015.6343. Epub 2016 Feb 1.
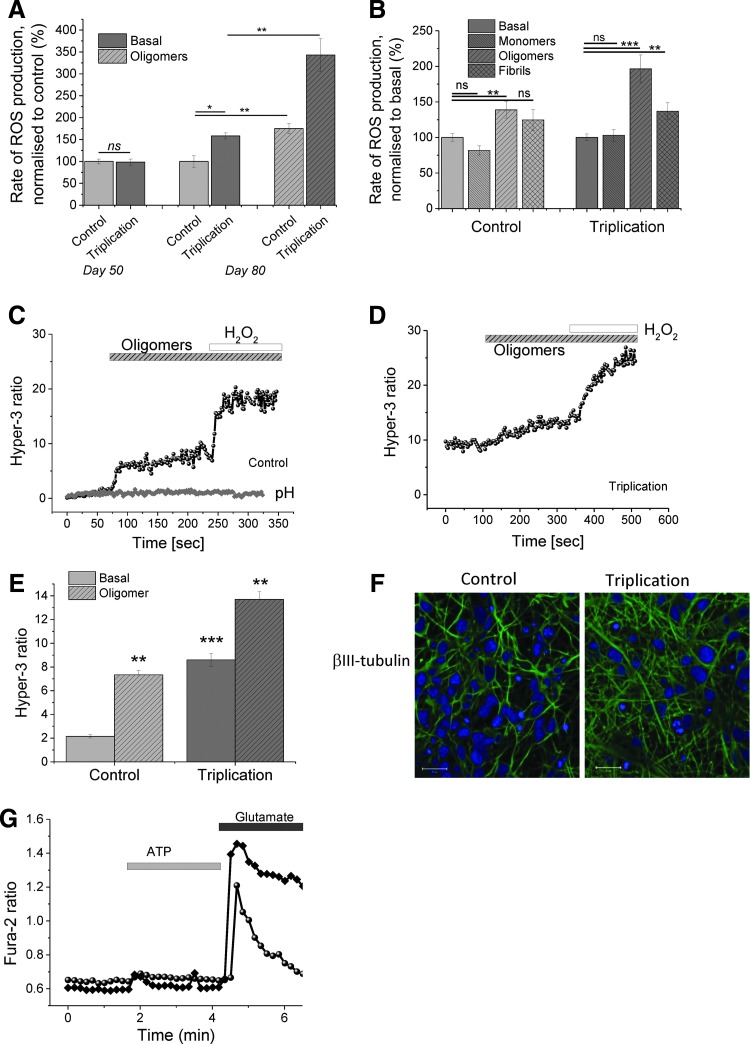
Protein aggregation and oxidative stress are both key pathogenic processes in Parkinson's disease, although the mechanism by which misfolded proteins induce oxidative stress and neuronal death remains unknown. In this study, we describe how aggregation of alpha-synuclein (α-S) from its monomeric form to its soluble oligomeric state results in aberrant free radical production and neuronal toxicity.
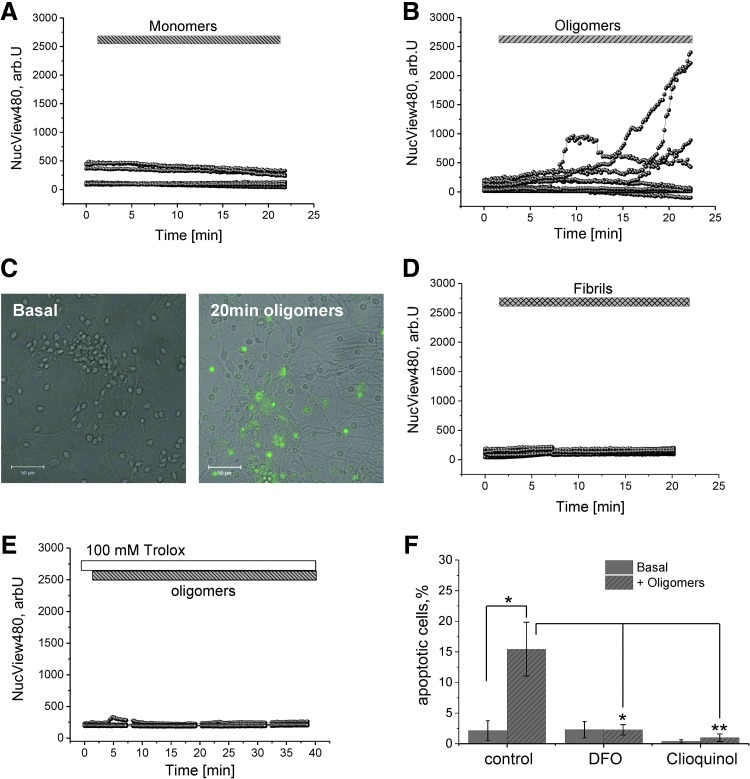
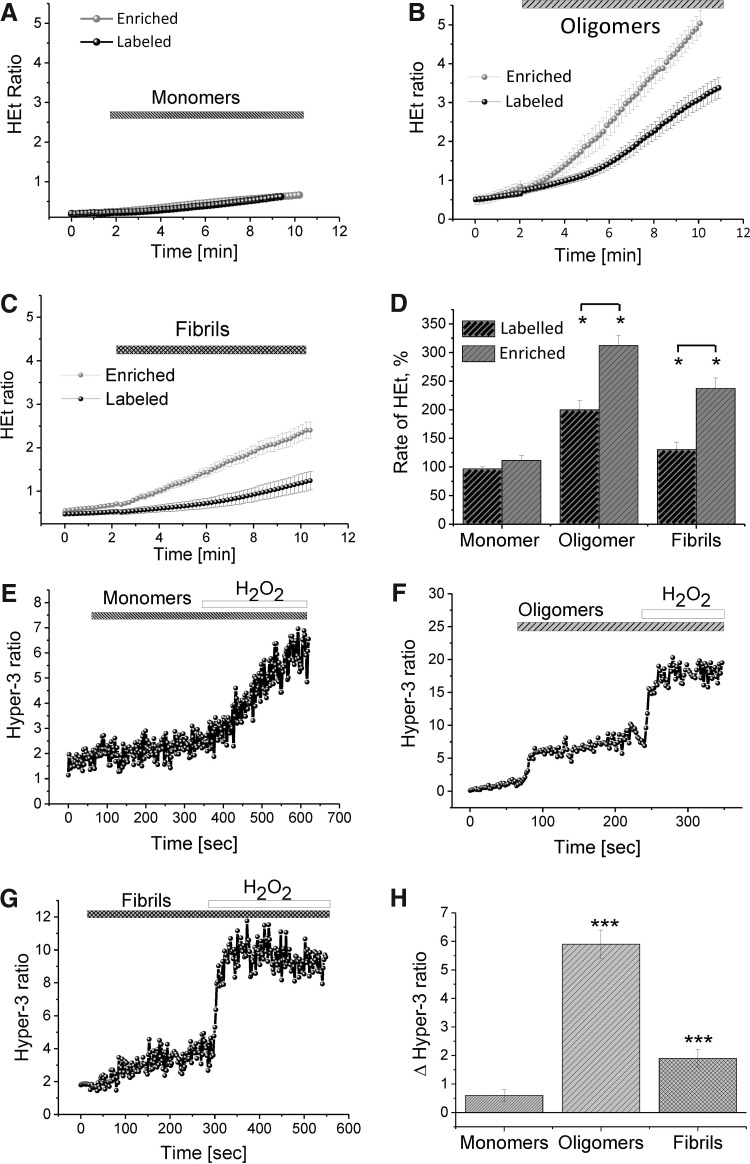
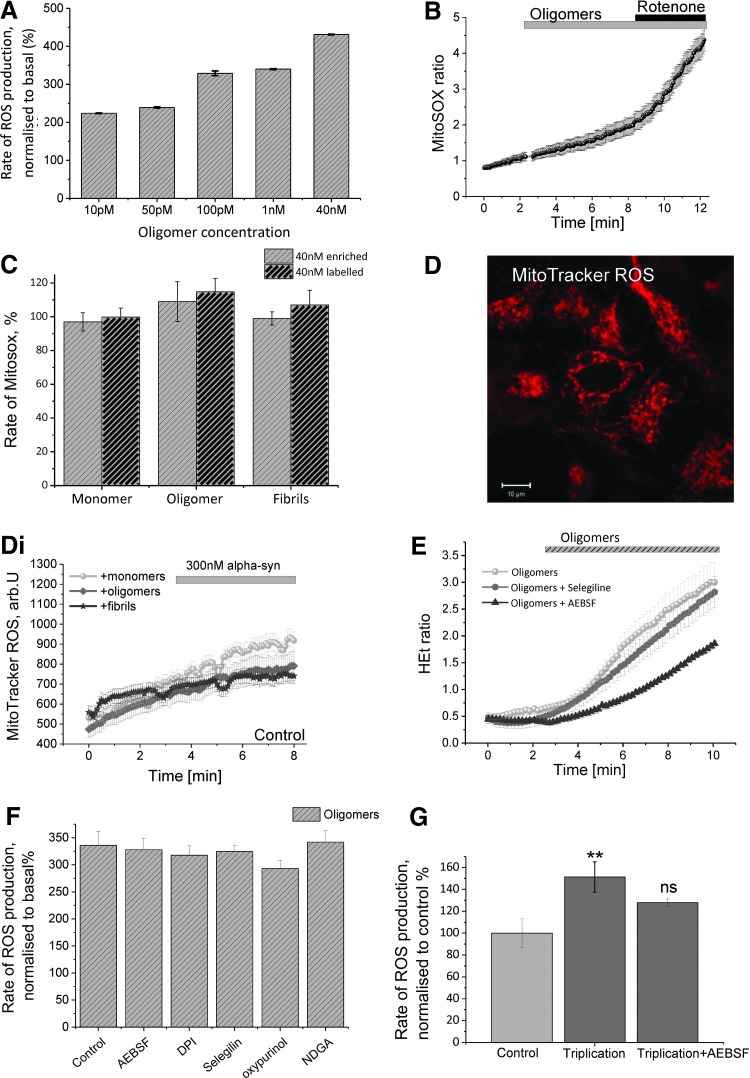
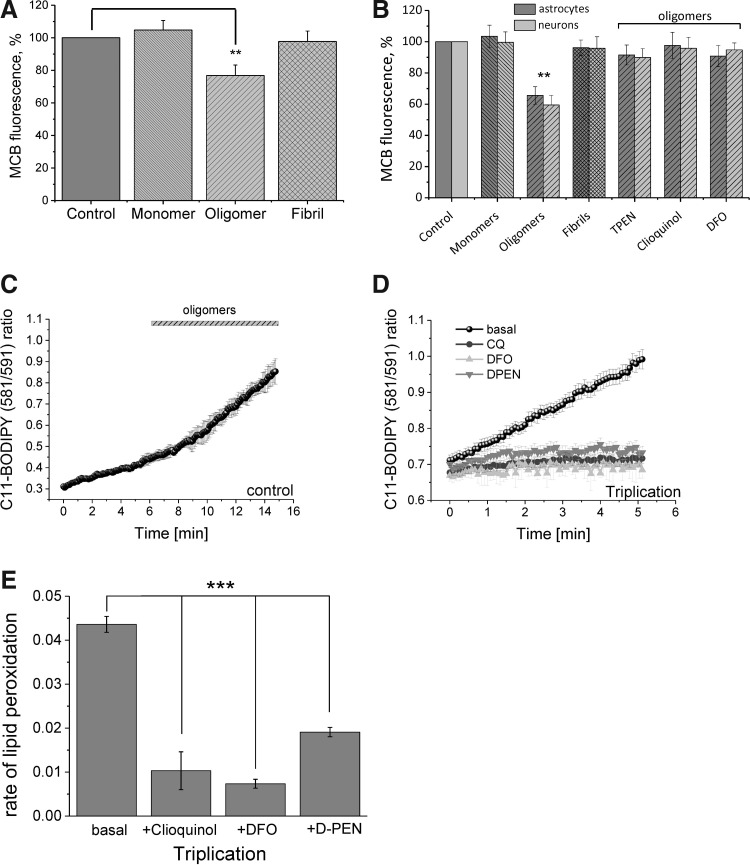
We first demonstrate excessive free radical production in a human induced pluripotent stem-derived α-S triplication model at basal levels and on application of picomolar doses of β-sheet-rich α-S oligomers. We probed the effects of different structural species of α-S in wild-type rat neuronal cultures and show that both oligomeric and fibrillar forms of α-S are capable of generating free radical production, but that only the oligomeric form results in reduction of endogenous glutathione and subsequent neuronal toxicity. We dissected the mechanism of oligomer-induced free radical production and found that it was interestingly independent of several known cellular enzymatic sources.
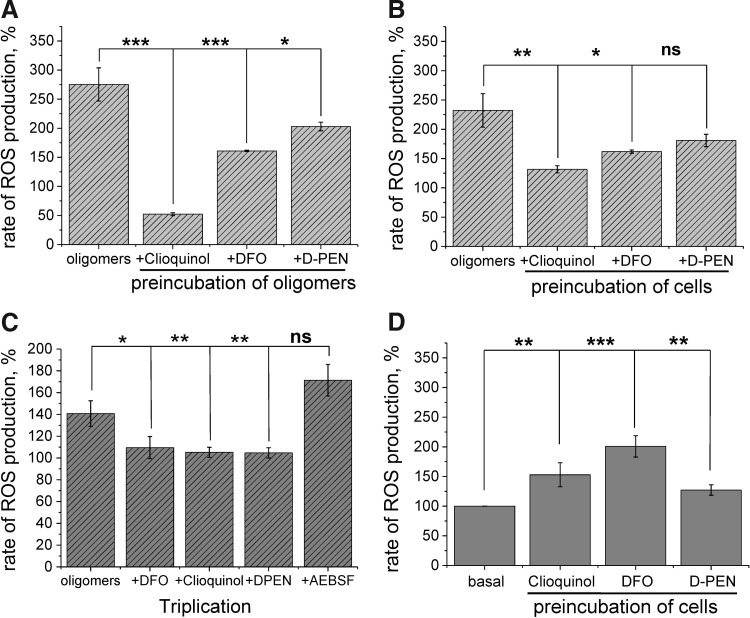
The oligomer-induced reactive oxygen species (ROS) production was entirely dependent on the presence of free metal ions as addition of metal chelators was able to block oligomer-induced ROS production and prevent oligomer-induced neuronal death.
Our findings further support the causative role of soluble amyloid oligomers in triggering neurodegeneration and shed light into the mechanisms by which these species cause neuronal damage, which, we show here, can be amenable to modulation through the use of metal chelation.
蛋白质聚集和氧化应激都是帕金森病的关键致病过程,尽管错误折叠的蛋白质诱导氧化应激和神经元死亡的机制尚不清楚。在本研究中,我们描述了α-突触核蛋白(α-S)从单体形式聚集到可溶性寡聚状态如何导致异常的自由基产生和神经元毒性。
我们首先在人诱导多能干细胞衍生的α-S三倍体模型中,在基础水平以及应用皮摩尔剂量的富含β-折叠的α-S寡聚体时,证明了过量的自由基产生。我们在野生型大鼠神经元培养物中探究了不同结构形式的α-S的作用,结果表明α-S的寡聚体形式和纤维状形式都能够产生自由基,但只有寡聚体形式会导致内源性谷胱甘肽减少及随后的神经元毒性。我们剖析了寡聚体诱导自由基产生的机制,发现有趣的是,它独立于几种已知的细胞酶源。
寡聚体诱导的活性氧(ROS)产生完全依赖于游离金属离子的存在,因为添加金属螯合剂能够阻断寡聚体诱导的ROS产生并防止寡聚体诱导的神经元死亡。
我们的研究结果进一步支持了可溶性淀粉样寡聚体在引发神经退行性变中的致病作用,并揭示了这些物质导致神经元损伤的机制,我们在此表明,通过使用金属螯合作用可以对其进行调节。