Audain Enrique, Ramos Yassel, Hermjakob Henning, Flower Darren R, Perez-Riverol Yasset
Department of Proteomics, Center of Molecular Immunology.
Department of Proteomics, Center for Genetic Engineering and Biotechnology, Ciudad de la Habana, Cuba.
Bioinformatics. 2016 Mar 15;32(6):821-7. doi: 10.1093/bioinformatics/btv674. Epub 2015 Nov 14.
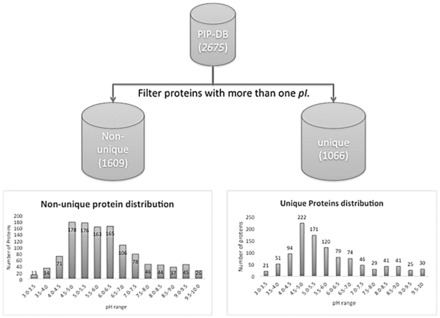
In any macromolecular polyprotic system-for example protein, DNA or RNA-the isoelectric point-commonly referred to as the pI-can be defined as the point of singularity in a titration curve, corresponding to the solution pH value at which the net overall surface charge-and thus the electrophoretic mobility-of the ampholyte sums to zero. Different modern analytical biochemistry and proteomics methods depend on the isoelectric point as a principal feature for protein and peptide characterization. Protein separation by isoelectric point is a critical part of 2-D gel electrophoresis, a key precursor of proteomics, where discrete spots can be digested in-gel, and proteins subsequently identified by analytical mass spectrometry. Peptide fractionation according to their pI is also widely used in current proteomics sample preparation procedures previous to the LC-MS/MS analysis. Therefore accurate theoretical prediction of pI would expedite such analysis. While such pI calculation is widely used, it remains largely untested, motivating our efforts to benchmark pI prediction methods.
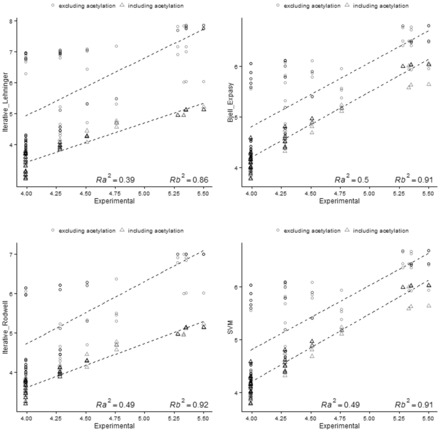
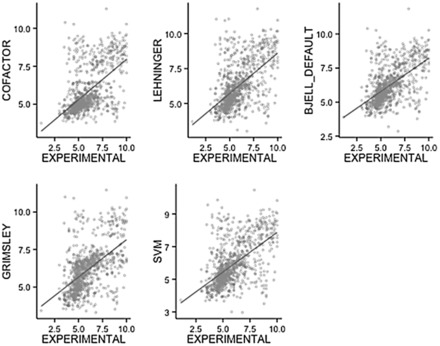
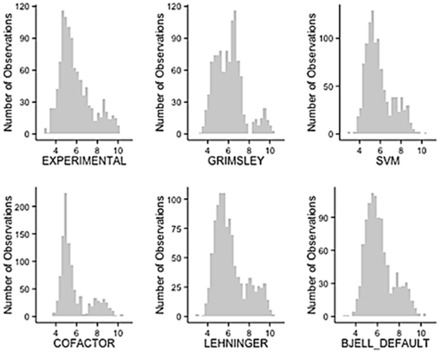
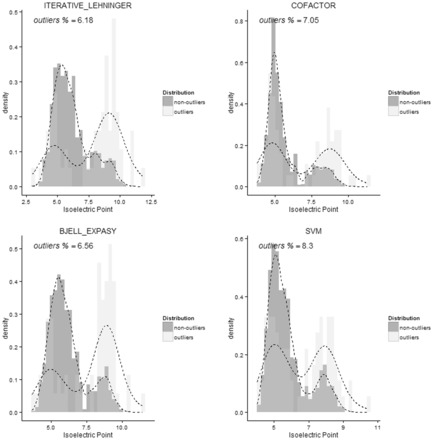
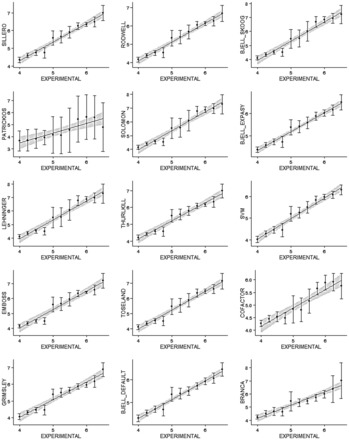
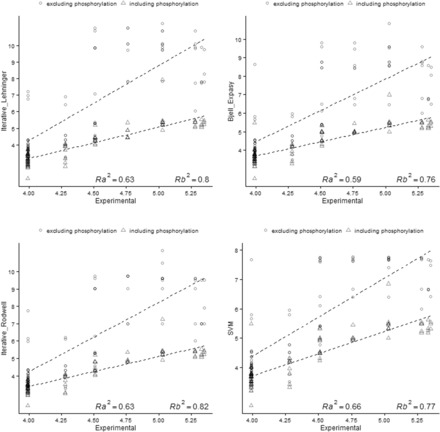
Using data from the database PIP-DB and one publically available dataset as our reference gold standard, we have undertaken the benchmarking of pI calculation methods. We find that methods vary in their accuracy and are highly sensitive to the choice of basis set. The machine-learning algorithms, especially the SVM-based algorithm, showed a superior performance when studying peptide mixtures. In general, learning-based pI prediction methods (such as Cofactor, SVM and Branca) require a large training dataset and their resulting performance will strongly depend of the quality of that data. In contrast with Iterative methods, machine-learning algorithms have the advantage of being able to add new features to improve the accuracy of prediction.
The software and data are freely available at https://github.com/ypriverol/pIRSupplementary information: Supplementary data are available at Bioinformatics online.
在任何大分子多质子体系中——例如蛋白质、DNA或RNA——等电点(通常称为pI)可定义为滴定曲线中的奇点,对应于两性电解质净总表面电荷(以及因此的电泳迁移率)总和为零的溶液pH值。不同的现代分析生物化学和蛋白质组学方法依赖等电点作为蛋白质和肽表征的主要特征。通过等电点进行蛋白质分离是二维凝胶电泳的关键部分,二维凝胶电泳是蛋白质组学的关键前身,其中离散的斑点可在凝胶内进行消化,随后通过分析质谱法鉴定蛋白质。根据肽的pI进行分级分离也广泛用于当前蛋白质组学样品制备程序中LC-MS/MS分析之前。因此,准确的pI理论预测将加快此类分析。虽然这种pI计算被广泛使用,但在很大程度上仍未经过测试,这促使我们努力对标pI预测方法。
使用来自数据库PIP-DB的数据和一个公开可用的数据集作为我们的参考金标准,我们对标了pI计算方法。我们发现这些方法的准确性各不相同,并且对基组的选择高度敏感。机器学习算法,尤其是基于支持向量机的算法,在研究肽混合物时表现出卓越的性能。一般来说,基于学习的pI预测方法(如Cofactor、支持向量机和布兰卡)需要大量的训练数据集,其最终性能将强烈依赖于该数据的质量。与迭代方法相比,机器学习算法的优势在于能够添加新特征以提高预测准确性。
软件和数据可在https://github.com/ypriverol/pI免费获取。补充信息:补充数据可在《生物信息学》在线获取。