Gobbini Elisa, Villa Matteo, Gnugnoli Marco, Menin Luca, Clerici Michela, Longhese Maria Pia
Dipartimento di Biotecnologie e Bioscienze, Università di Milano-Bicocca, Milano, Italy.
PLoS Genet. 2015 Nov 19;11(11):e1005685. doi: 10.1371/journal.pgen.1005685. eCollection 2015 Nov.
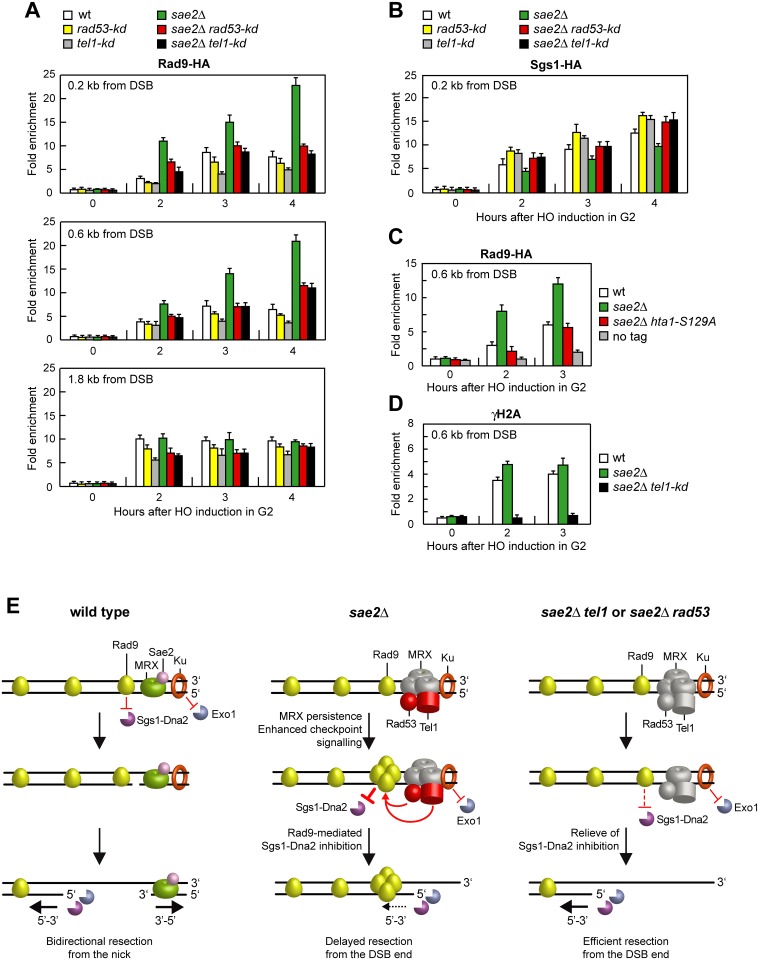
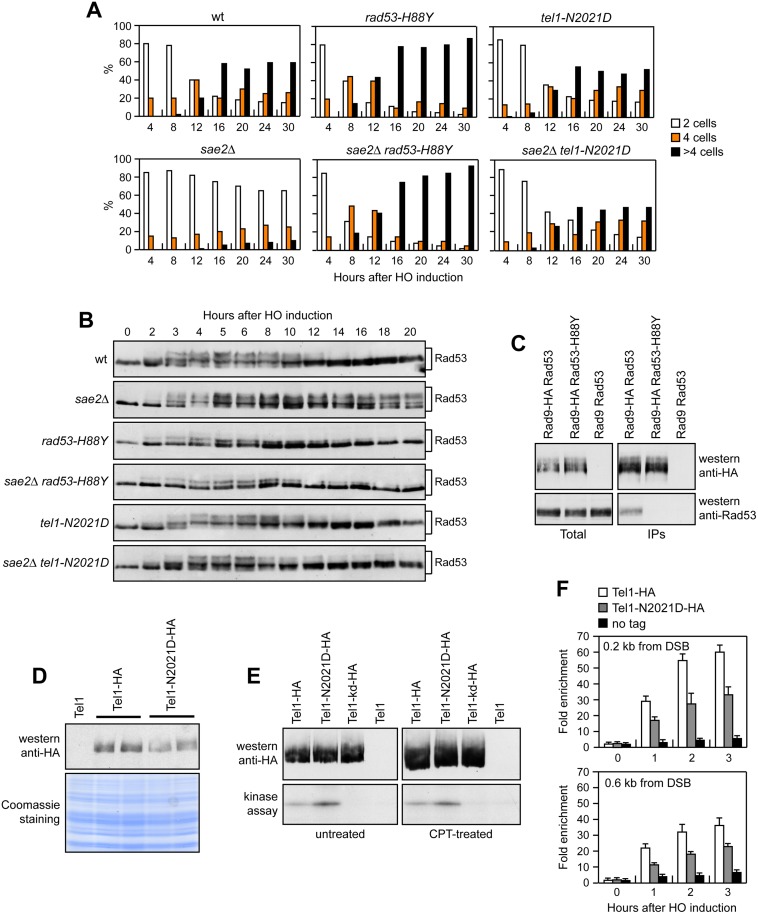
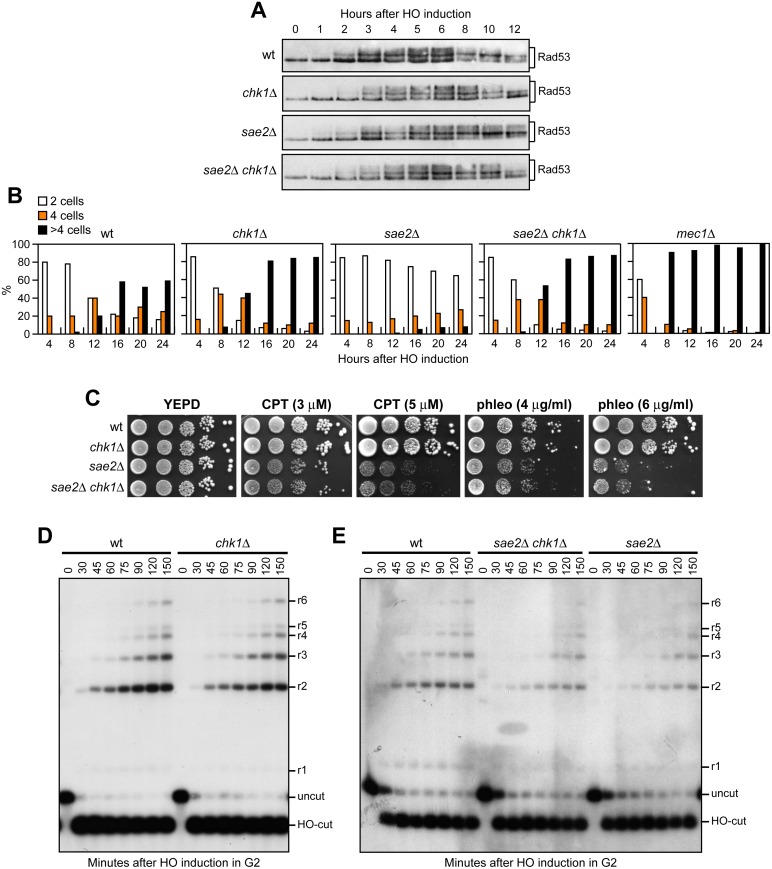
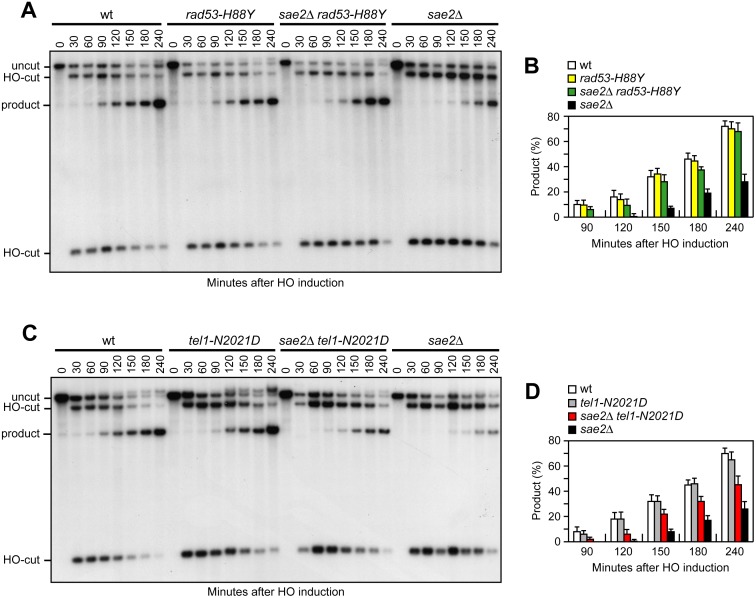
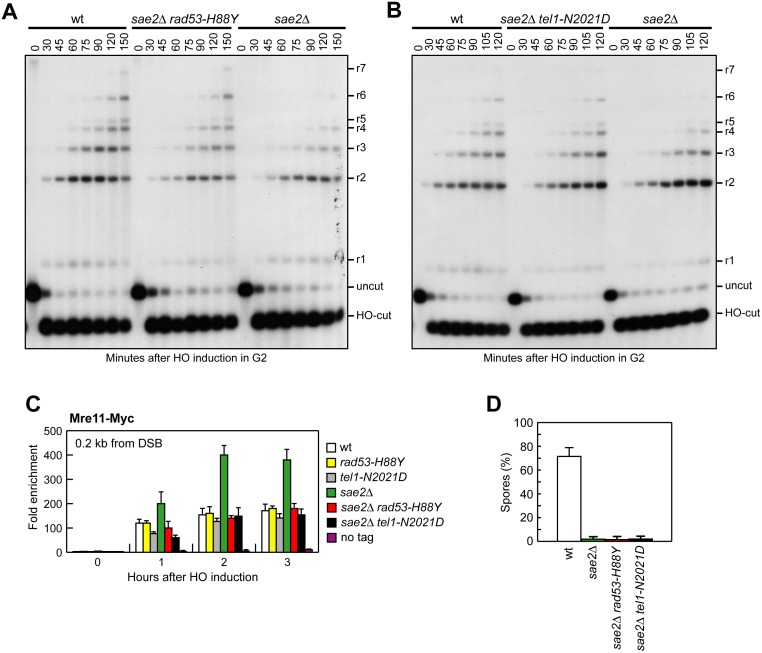
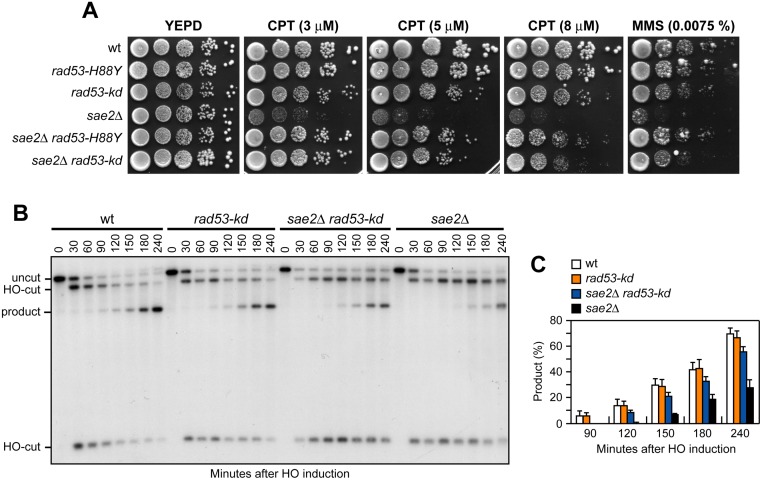
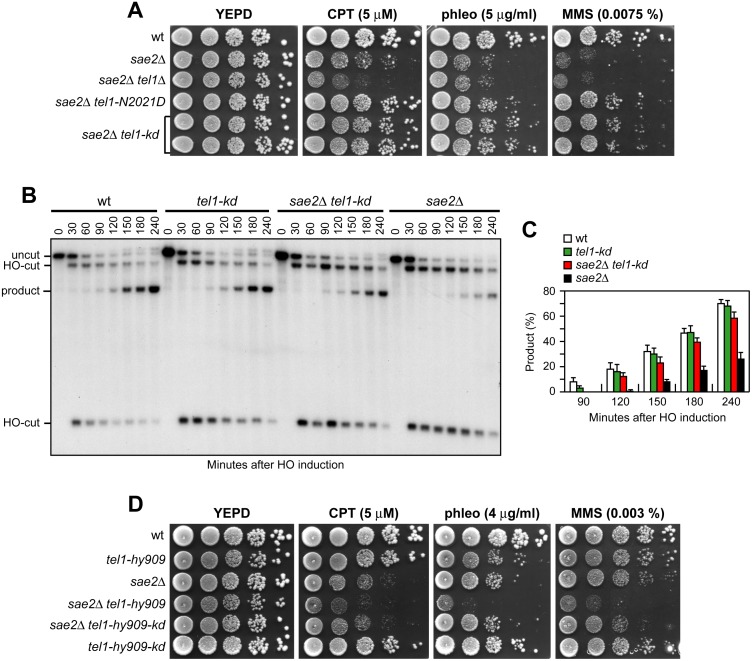
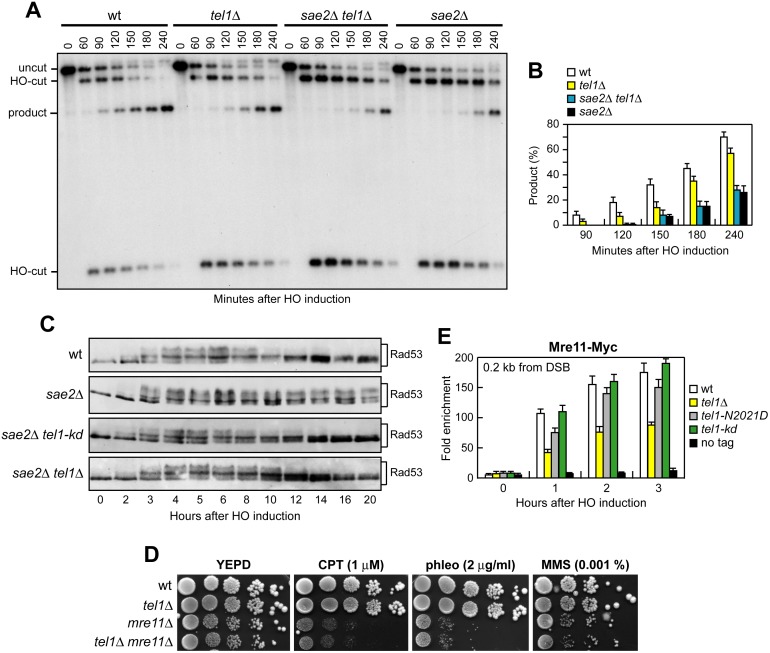
The MRX complex together with Sae2 initiates resection of DNA double-strand breaks (DSBs) to generate single-stranded DNA (ssDNA) that triggers homologous recombination. The absence of Sae2 not only impairs DSB resection, but also causes prolonged MRX binding at the DSBs that leads to persistent Tel1- and Rad53-dependent DNA damage checkpoint activation and cell cycle arrest. Whether this enhanced checkpoint signaling contributes to the DNA damage sensitivity and/or the resection defect of sae2Δ cells is not known. By performing a genetic screen, we identify rad53 and tel1 mutant alleles that suppress both the DNA damage hypersensitivity and the resection defect of sae2Δ cells through an Sgs1-Dna2-dependent mechanism. These suppression events do not involve escaping the checkpoint-mediated cell cycle arrest. Rather, defective Rad53 or Tel1 signaling bypasses Sae2 function at DSBs by decreasing the amount of Rad9 bound at DSBs. As a consequence, reduced Rad9 association to DNA ends relieves inhibition of Sgs1-Dna2 activity, which can then compensate for the lack of Sae2 in DSB resection and DNA damage resistance. We propose that persistent Tel1 and Rad53 checkpoint signaling in cells lacking Sae2 increases the association of Rad9 at DSBs, which in turn inhibits DSB resection by limiting the activity of the Sgs1-Dna2 resection machinery.
MRX复合物与Sae2共同启动DNA双链断裂(DSB)的切除,以生成触发同源重组的单链DNA(ssDNA)。Sae2的缺失不仅会损害DSB切除,还会导致MRX在DSB处的结合时间延长,从而导致持续的Tel1和Rad53依赖性DNA损伤检查点激活和细胞周期停滞。这种增强的检查点信号是否导致sae2Δ细胞的DNA损伤敏感性和/或切除缺陷尚不清楚。通过进行遗传筛选,我们鉴定出rad53和tel1突变等位基因,它们通过Sgs1-Dna2依赖性机制抑制sae2Δ细胞的DNA损伤超敏性和切除缺陷。这些抑制事件不涉及逃避检查点介导的细胞周期停滞。相反,有缺陷的Rad53或Tel1信号传导通过减少DSB处结合的Rad9量来绕过DSB处的Sae2功能。因此,Rad9与DNA末端的结合减少缓解了对Sgs1-Dna2活性的抑制,从而可以补偿DSB切除和DNA损伤抗性中Sae2的缺乏。我们提出,缺乏Sae2的细胞中持续的Tel1和Rad53检查点信号增加了Rad9在DSB处的结合,这反过来又通过限制Sgs1-Dna2切除机制的活性来抑制DSB切除。