Suyama Yoshihisa, Matsuki Yu
Tohoku University, Kawatabi Field Science Center, Graduate School of Agricultural Science, 232-3 Yomogida, Naruko-onsen, Osaki, Miyagi 989-6711, Japan.
Sci Rep. 2015 Nov 23;5:16963. doi: 10.1038/srep16963.
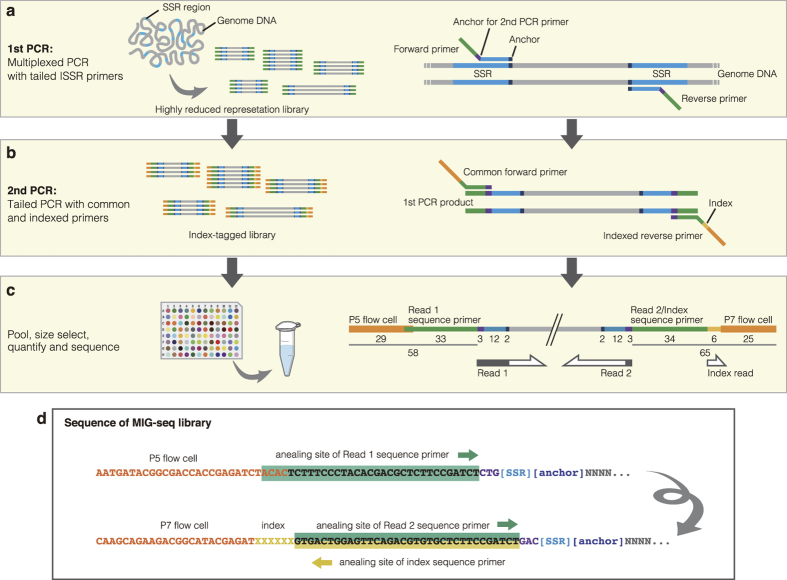
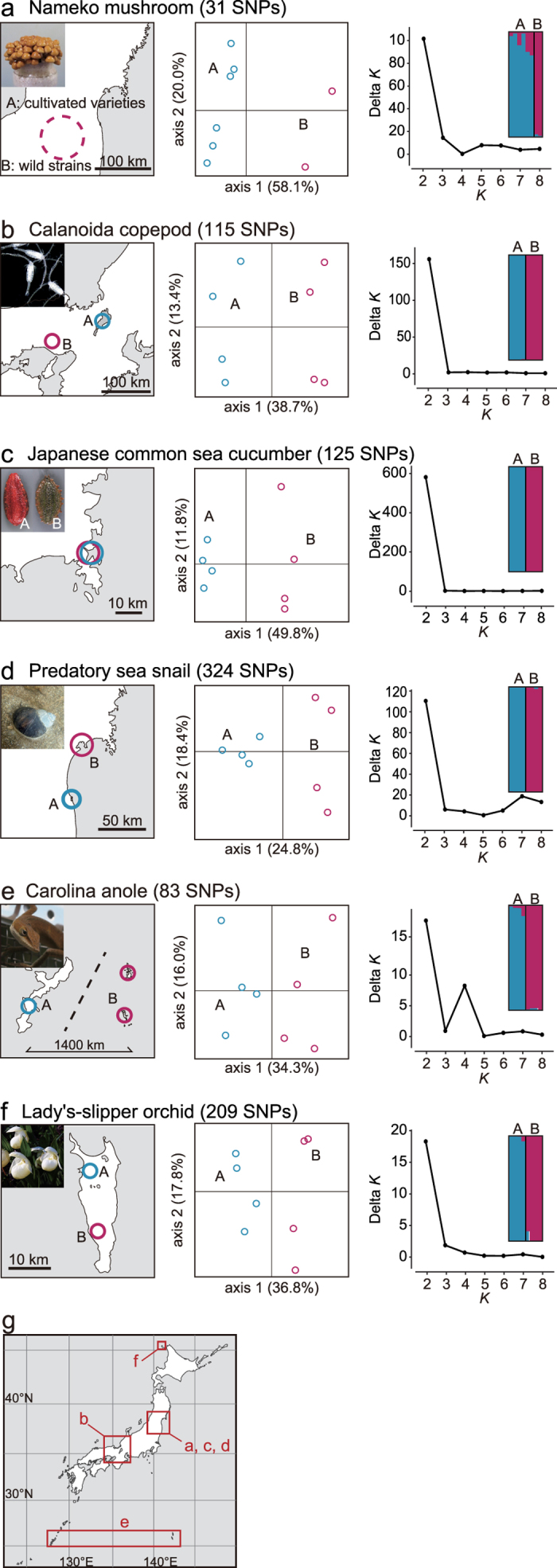
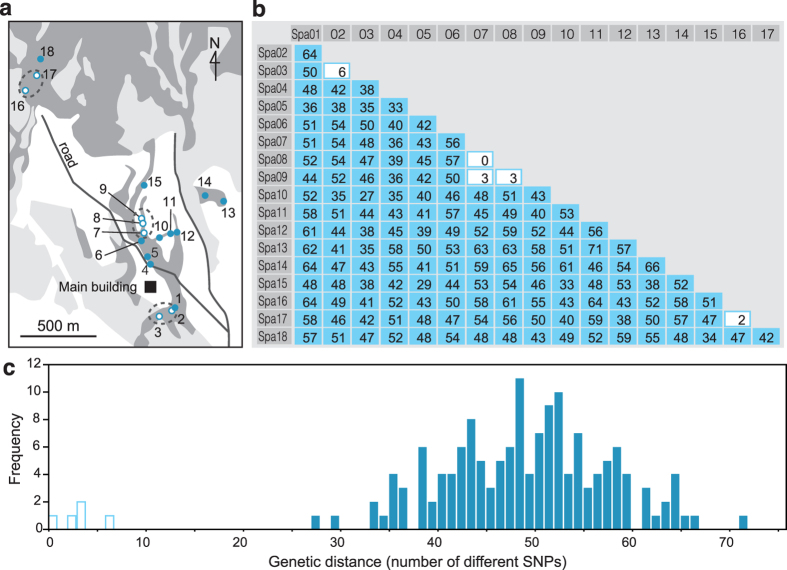
Restriction-enzyme (RE)-based next-generation sequencing methods have revolutionized marker-assisted genetic studies; however, the use of REs has limited their widespread adoption, especially in field samples with low-quality DNA and/or small quantities of DNA. Here, we developed a PCR-based procedure to construct reduced representation libraries without RE digestion steps, representing de novo single-nucleotide polymorphism discovery, and its genotyping using next-generation sequencing. Using multiplexed inter-simple sequence repeat (ISSR) primers, thousands of genome-wide regions were amplified effectively from a wide variety of genomes, without prior genetic information. We demonstrated: 1) Mendelian gametic segregation of the discovered variants; 2) reproducibility of genotyping by checking its applicability for individual identification; and 3) applicability in a wide variety of species by checking standard population genetic analysis. This approach, called multiplexed ISSR genotyping by sequencing, should be applicable to many marker-assisted genetic studies with a wide range of DNA qualities and quantities.
基于限制性内切酶(RE)的新一代测序方法彻底改变了标记辅助遗传研究;然而,限制性内切酶的使用限制了它们的广泛应用,尤其是在DNA质量低和/或DNA数量少的野外样本中。在此,我们开发了一种基于PCR的程序,用于构建简化代表性文库,无需限制性内切酶消化步骤,可实现从头单核苷酸多态性发现及其利用新一代测序进行基因分型。使用多重简单序列重复区间(ISSR)引物,无需先验遗传信息,就能从多种基因组中有效扩增数千个全基因组区域。我们证明了:1)所发现变异的孟德尔配子分离;2)通过检查其在个体识别中的适用性来验证基因分型的可重复性;3)通过检查标准群体遗传分析来验证其在多种物种中的适用性。这种方法称为多重ISSR测序基因分型,应该适用于许多具有广泛DNA质量和数量的标记辅助遗传研究。