Caro-Quintero Alejandro, Ochman Howard
Department of Integrative Biology, University of Texas, Austin Present address: Corpoicá C.I Tibaitata, Santáfe de Bogata, Columbia.
Department of Integrative Biology, University of Texas, Austin
Genome Biol Evol. 2015 Nov 27;7(12):3416-25. doi: 10.1093/gbe/evv234.
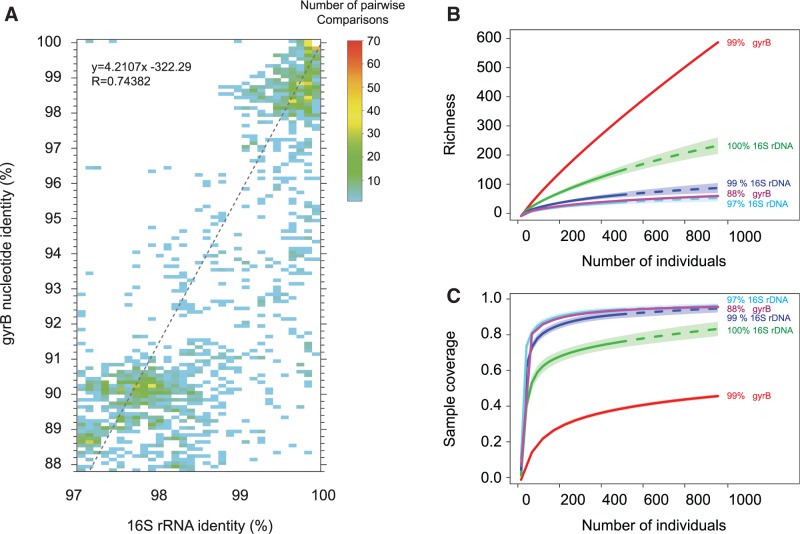
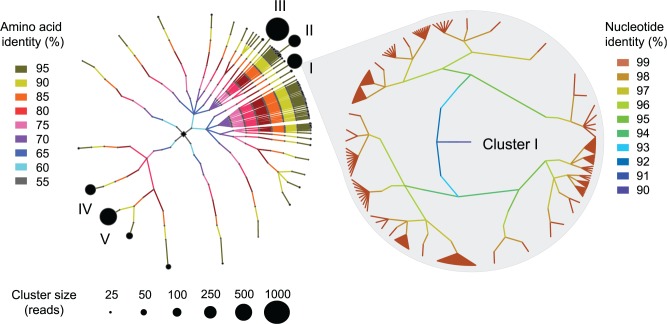
For both historical and technical reasons, 16S ribosomal RNA has been the most common molecular marker used to analyze the contents of microbial communities. However, its slow rate of evolution hinders the resolution of closely related bacteria--individual 16S-phylotypes, particularly when clustered at 97% sequence identity, conceal vast amounts of species- and strain-level variation. Protein-coding genes, which evolve more quickly, are useful for differentiating among more recently diverged lineages, but their application is complicated by difficulties in designing low-redundancy primers that amplify homologous regions from distantly related taxa. Given the now-common practice of multiplexing hundreds of samples, adopting new genes usually entails the synthesis of large sets of barcoded primers. To circumvent problems associated with use of protein-coding genes to survey microbial communities, we develop an approach--termed phyloTAGs--that offers an automatic solution for primer design and can be easily adapted to target different taxonomic groups and/or different protein-coding regions. We applied this method to analyze diversity within the gorilla gut microbiome and recovered hundreds of strains that went undetected after deep-sequencing of 16S rDNA amplicons. PhyloTAGs provides a powerful way to recover the fine-level diversity within microbial communities and to study stability and dynamics of bacterial populations.
由于历史和技术原因,16S核糖体RNA一直是用于分析微生物群落组成的最常用分子标记。然而,其缓慢的进化速度阻碍了对亲缘关系较近的细菌的分辨——单个16S系统型,尤其是当以97%的序列同一性聚类时,掩盖了大量物种和菌株水平的变异。进化速度更快的蛋白质编码基因,对于区分最近分化的谱系很有用,但由于难以设计出能从远缘分类群中扩增同源区域的低冗余引物,其应用变得复杂。鉴于现在对数百个样本进行多重分析的普遍做法,采用新基因通常需要合成大量带条形码的引物。为了规避使用蛋白质编码基因来调查微生物群落所带来的问题,我们开发了一种方法——称为系统发育标签法(phyloTAGs)——它为引物设计提供了一种自动解决方案,并且可以很容易地适用于针对不同的分类群和/或不同的蛋白质编码区域。我们应用这种方法来分析大猩猩肠道微生物组内的多样性,并发现了数百个在16S rDNA扩增子深度测序后未被检测到的菌株。系统发育标签法提供了一种强大的方法来恢复微生物群落内的精细水平多样性,并研究细菌种群的稳定性和动态。