Chesi Giancarlo, Hegde Ramanath N, Iacobacci Simona, Concilli Mafalda, Parashuraman Seetharaman, Festa Beatrice Paola, Polishchuk Elena V, Di Tullio Giuseppe, Carissimo Annamaria, Montefusco Sandro, Canetti Diana, Monti Maria, Amoresano Angela, Pucci Piero, van de Sluis Bart, Lutsenko Svetlana, Luini Alberto, Polishchuk Roman S
Telethon Institute of Genetics and Medicine, Pozzuoli, Italy.
Institute of Protein Biochemistry, National Research Council, Naples, Italy.
Hepatology. 2016 Jun;63(6):1842-59. doi: 10.1002/hep.28398. Epub 2016 Jan 25.
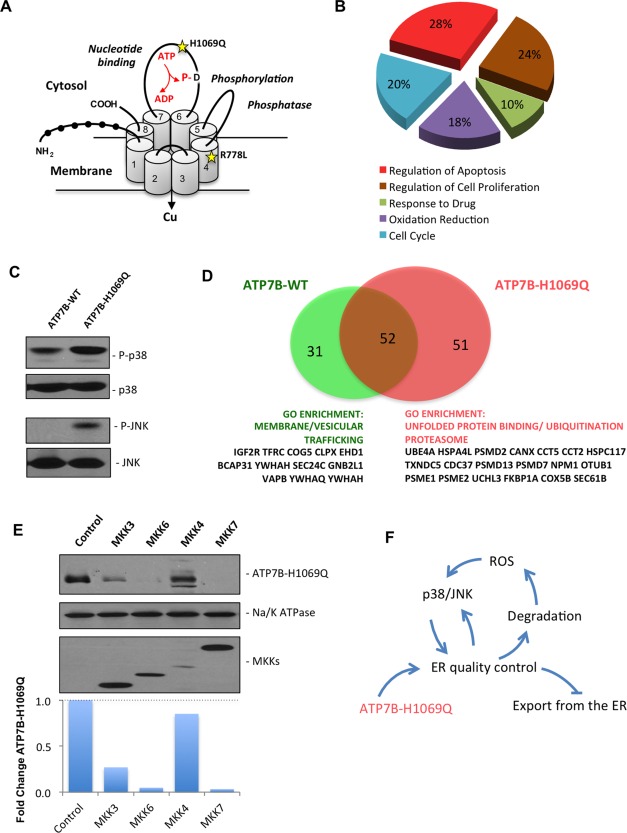
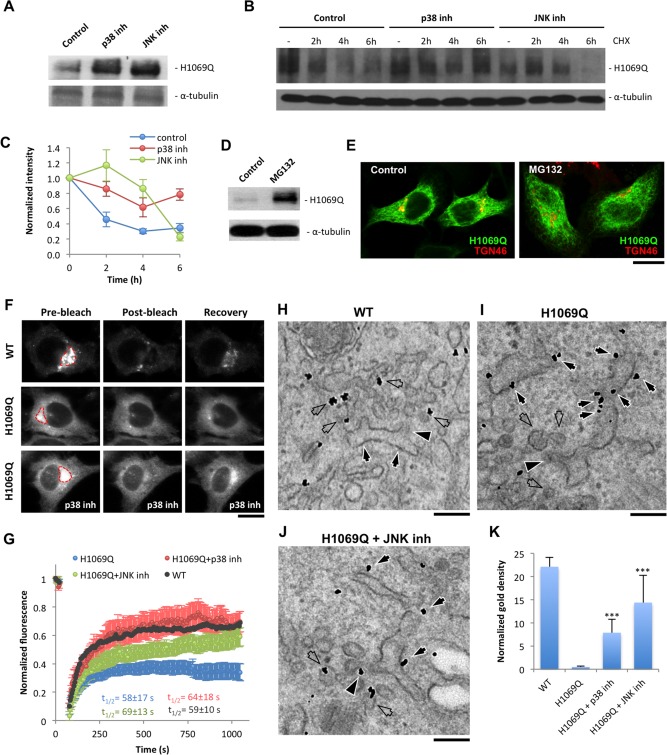
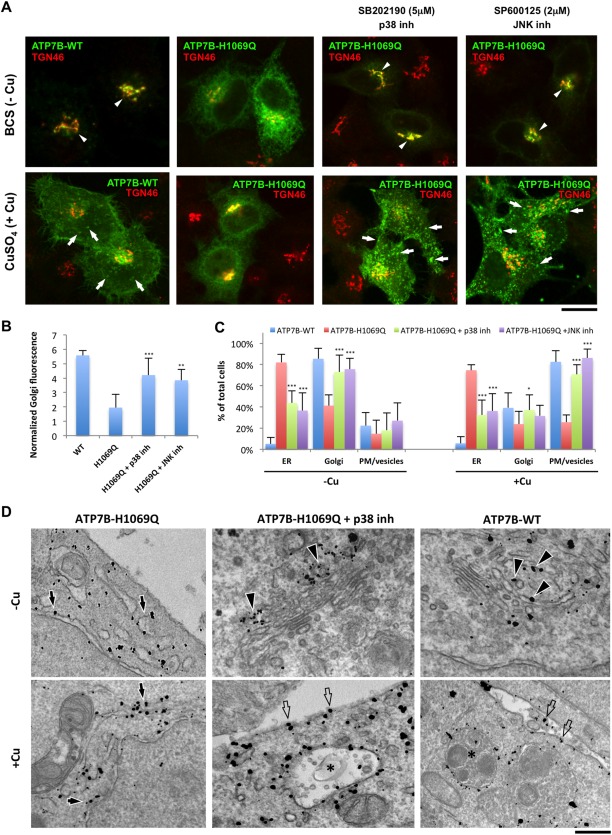
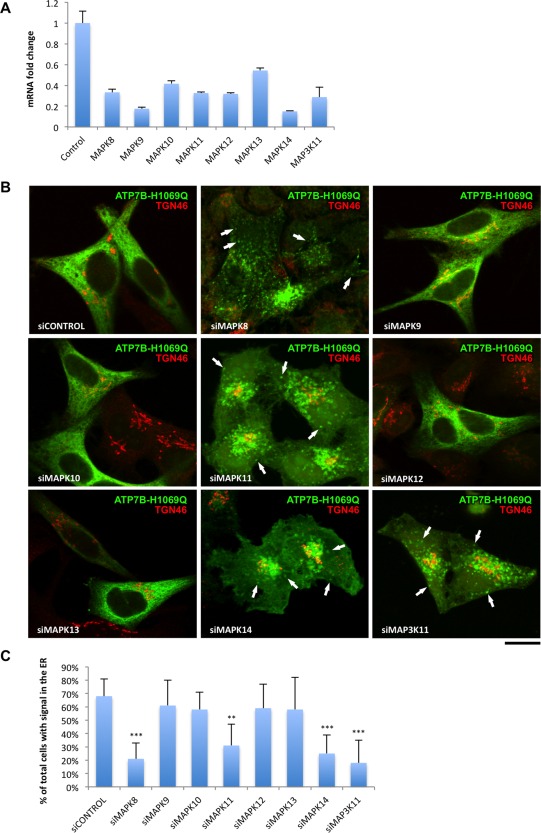
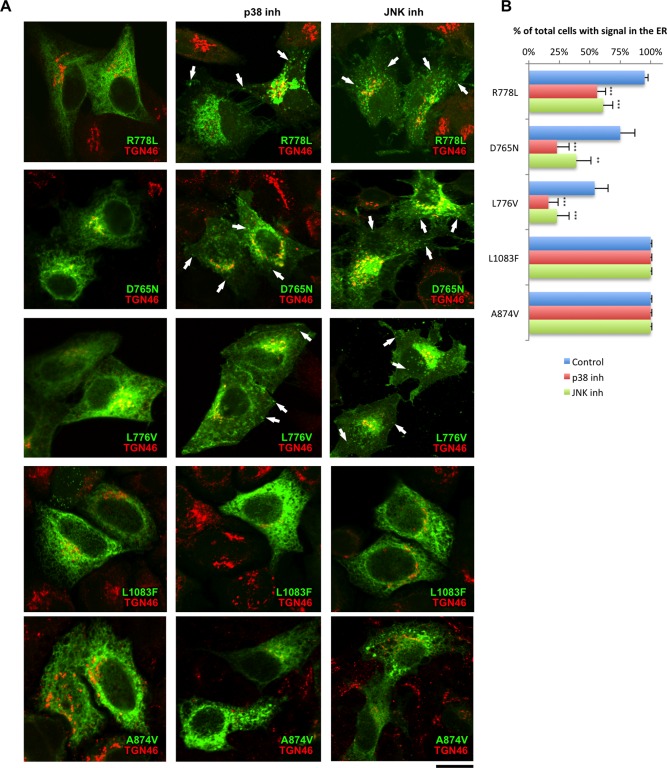
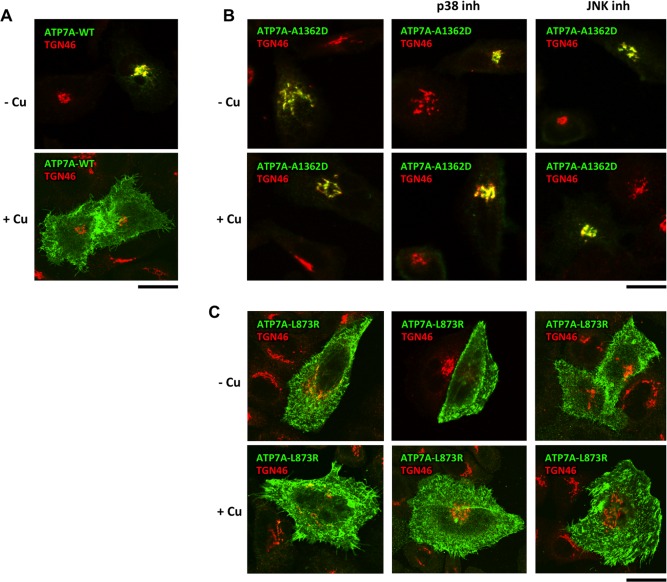
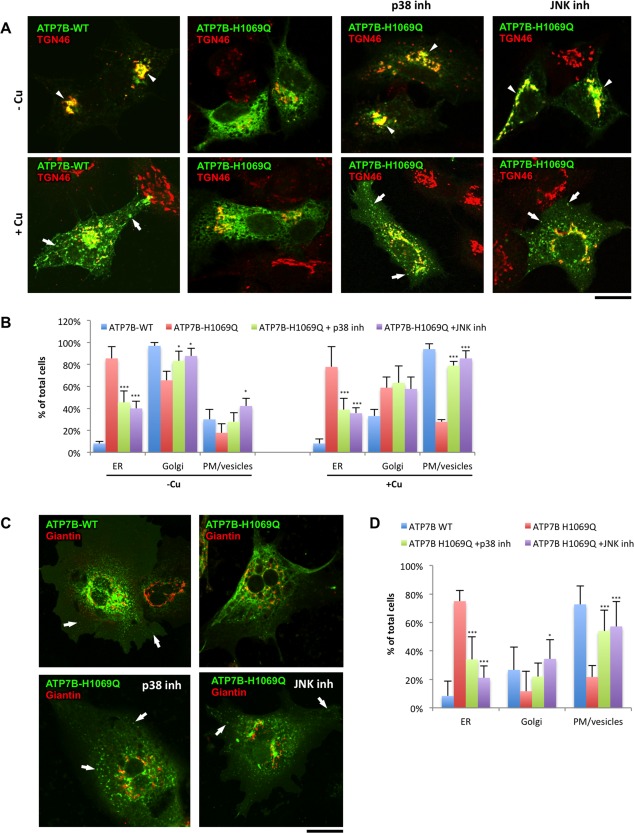
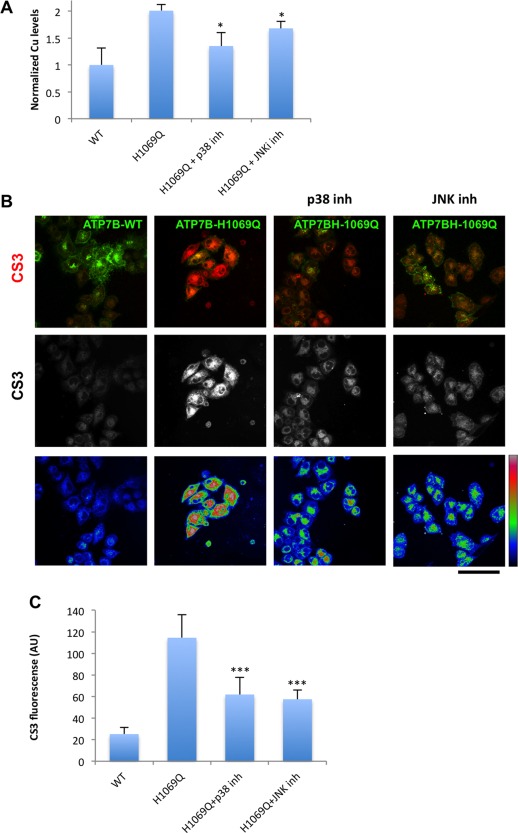
Wilson disease (WD) is an autosomal recessive disorder that is caused by the toxic accumulation of copper (Cu) in the liver. The ATP7B gene, which is mutated in WD, encodes a multitransmembrane domain adenosine triphosphatase that traffics from the trans-Golgi network to the canalicular area of hepatocytes, where it facilitates excretion of excess Cu into the bile. Several ATP7B mutations, including H1069Q and R778L that are two of the most frequent variants, result in protein products, which, although still functional, remain in the endoplasmic reticulum. Thus, they fail to reach Cu excretion sites, resulting in the toxic buildup of Cu in the liver of WD patients. Therefore, correcting the location of these mutants by leading them to the appropriate functional sites in the cell should restore Cu excretion and would be beneficial to help large cohorts of WD patients. However, molecular targets for correction of endoplasmic reticulum-retained ATP7B mutants remain elusive. Here, we show that expression of the most frequent ATP7B mutant, H1069Q, activates p38 and c-Jun N-terminal kinase signaling pathways, which favor the rapid degradation of the mutant. Suppression of these pathways with RNA interference or specific chemical inhibitors results in the substantial rescue of ATP7B(H1069Q) (as well as that of several other WD-causing mutants) from the endoplasmic reticulum to the trans-Golgi network compartment, in recovery of its Cu-dependent trafficking, and in reduction of intracellular Cu levels.
Our findings indicate p38 and c-Jun N-terminal kinase as intriguing targets for correction of WD-causing mutants and, hence, as potential candidates, which could be evaluated for the development of novel therapeutic strategies to combat WD. (Hepatology 2016;63:1842-1859).
威尔逊病(WD)是一种常染色体隐性疾病,由肝脏中铜(Cu)的毒性蓄积所致。WD中发生突变的ATP7B基因编码一种多跨膜结构域腺苷三磷酸酶,该酶从反式高尔基体网络转运至肝细胞的胆小管区域,在那里促进多余的铜排泄到胆汁中。几种ATP7B突变,包括最常见的两种变体H1069Q和R778L,会产生蛋白质产物,这些产物虽然仍有功能,但会滞留在内质网中。因此,它们无法到达铜排泄位点,导致WD患者肝脏中铜的毒性蓄积。所以,通过引导这些突变体到达细胞内合适的功能位点来纠正其位置,应能恢复铜的排泄,对大量WD患者有益。然而,纠正内质网滞留的ATP7B突变体的分子靶点仍不明确。在此,我们表明最常见的ATP7B突变体H1069Q的表达激活了p38和c-Jun氨基末端激酶信号通路,这有利于该突变体的快速降解。用RNA干扰或特异性化学抑制剂抑制这些通路,可使ATP7B(H1069Q)(以及其他几种导致WD的突变体)从内质网大量挽救至反式高尔基体网络区室,恢复其铜依赖性转运,并降低细胞内铜水平。
我们的研究结果表明p38和c-Jun氨基末端激酶是纠正导致WD的突变体的有趣靶点,因此作为潜在候选者,可对其进行评估以开发对抗WD的新型治疗策略。(《肝脏病学》2016年;63:1842 - 1859)