Olorin Emily, O'Brien Kevin T, Palopoli Nicolas, Pérez-Bercoff Åsa, Shields Denis C, Edwards Richard J
School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, Australia.
UCD Conway Institute of Biomolecular and Biomedical Research, School of Medicine, University College Dublin, Dublin, Ireland.
F1000Res. 2015 Aug 5;4:477. doi: 10.12688/f1000research.6773.1. eCollection 2015.
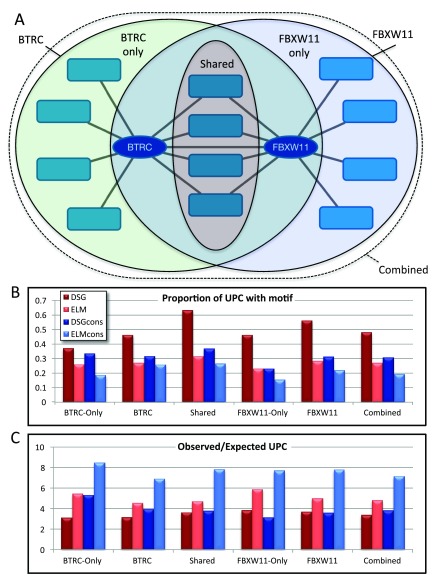
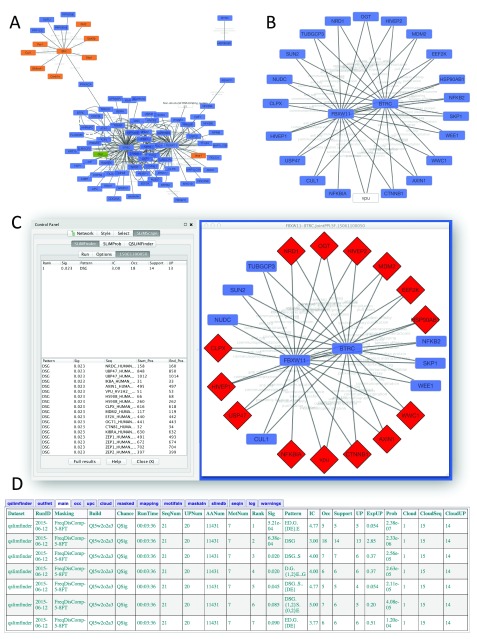
Short linear motifs (SLiMs) are small protein sequence patterns that mediate a large number of critical protein-protein interactions, involved in processes such as complex formation, signal transduction, localisation and stabilisation. SLiMs show rapid evolutionary dynamics and are frequently the targets of molecular mimicry by pathogens. Identifying enriched sequence patterns due to convergent evolution in non-homologous proteins has proven to be a successful strategy for computational SLiM prediction. Tools of the SLiMSuite package use this strategy, using a statistical model to identify SLiM enrichment based on the evolutionary relationships, amino acid composition and predicted disorder of the input proteins. The quality of input data is critical for successful SLiM prediction. Cytoscape provides a user-friendly, interactive environment to explore interaction networks and select proteins based on common features, such as shared interaction partners. SLiMScape embeds tools of the SLiMSuite package for de novo SLiM discovery (SLiMFinder and QSLiMFinder) and identifying occurrences/enrichment of known SLiMs (SLiMProb) within this interactive framework. SLiMScape makes it easier to (1) generate high quality hypothesis-driven datasets for these tools, and (2) visualise predicted SLiM occurrences within the context of the network. To generate new predictions, users can select nodes from a protein network or provide a set of Uniprot identifiers. SLiMProb also requires additional query motif input. Jobs are then run remotely on the SLiMSuite server ( http://rest.slimsuite.unsw.edu.au) for subsequent retrieval and visualisation. SLiMScape can also be used to retrieve and visualise results from jobs run directly on the server. SLiMScape and SLiMSuite are open source and freely available via GitHub under GNU licenses.
短线性基序(SLiMs)是介导大量关键蛋白质-蛋白质相互作用的小蛋白质序列模式,参与复合物形成、信号转导、定位和稳定等过程。SLiMs表现出快速的进化动态,并且经常是病原体分子模拟的目标。已证明识别非同源蛋白质中趋同进化导致的富集序列模式是计算SLiM预测的一种成功策略。SLiMSuite软件包的工具使用此策略,利用统计模型根据输入蛋白质的进化关系、氨基酸组成和预测的无序性来识别SLiM富集。输入数据的质量对于成功的SLiM预测至关重要。Cytoscape提供了一个用户友好的交互式环境,用于探索相互作用网络并根据共同特征(如共享的相互作用伙伴)选择蛋白质。SLiMScape在这个交互式框架中嵌入了SLiMsute软件包的工具,用于从头发现SLiM(SLiMFinder和QSLiMFinder)以及识别已知SLiM的出现/富集情况(SLiMProb)。SLiMScape使得更容易(1)为这些工具生成高质量的假设驱动数据集,以及(2)在网络背景下可视化预测的SLiM出现情况。为了生成新的预测,用户可以从蛋白质网络中选择节点或提供一组Uniprot标识符。SLiMProb还需要额外的查询基序输入。然后作业在SLiMSuite服务器(http://rest.slimsuite.unsw.edu.au)上远程运行,以便后续检索和可视化。SLiMScape还可用于检索和可视化直接在服务器上运行的作业的结果。SLiMScape和SLiMSuite是开源的,可通过GitHub在GNU许可下免费获得。