Idrees Sobia, Pérez-Bercoff Åsa, Edwards Richard J
School of Biotechnology and Biomolecular Sciences, University of New South Wales, Sydney, NSW, Australia.
PeerJ. 2018 Oct 31;6:e5858. doi: 10.7717/peerj.5858. eCollection 2018.
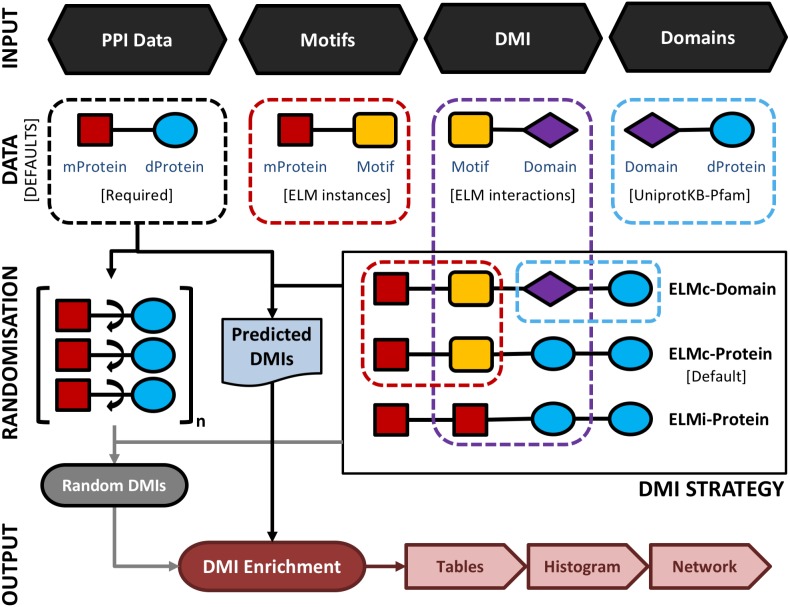
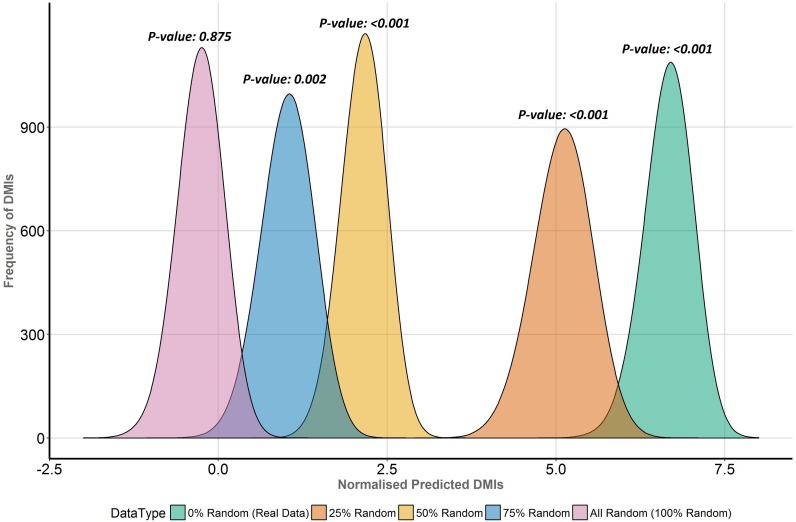
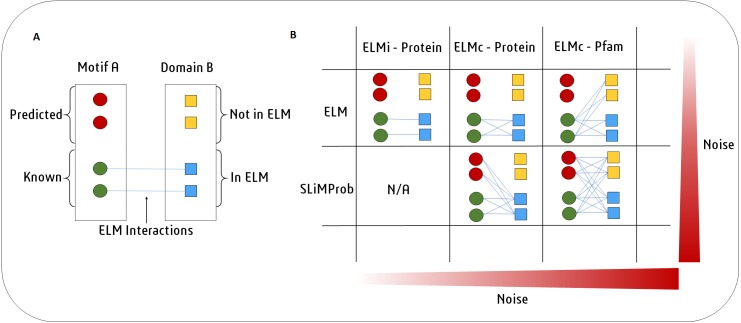
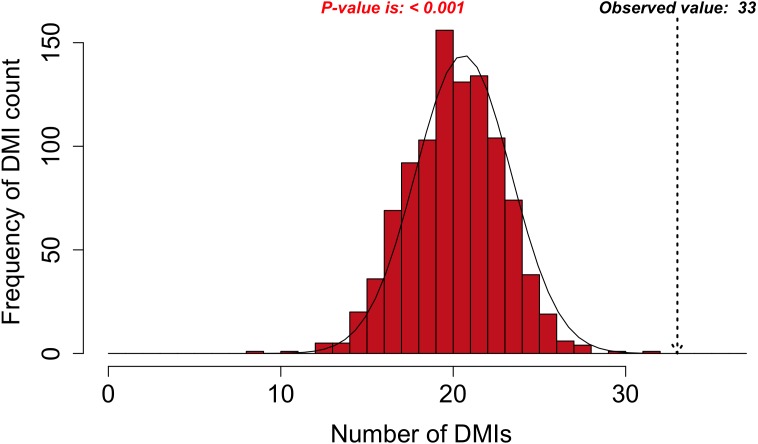
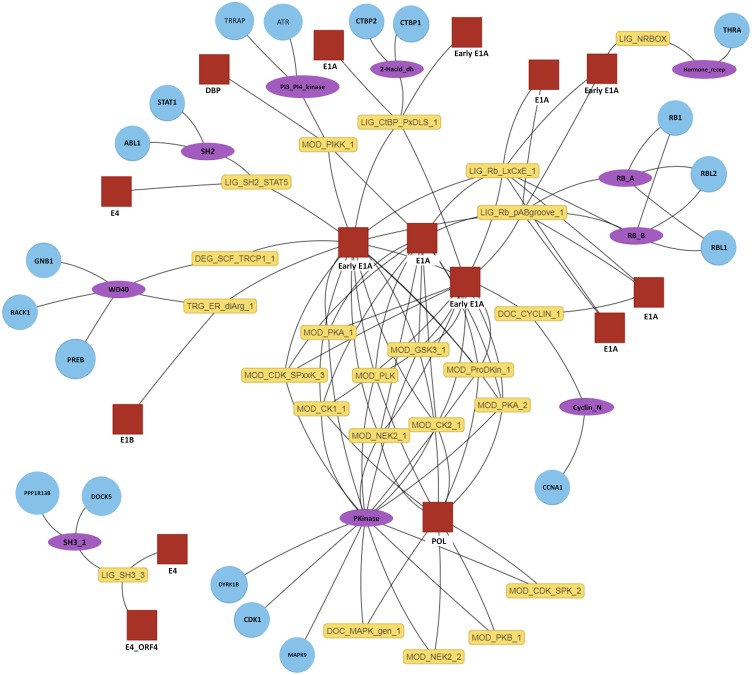
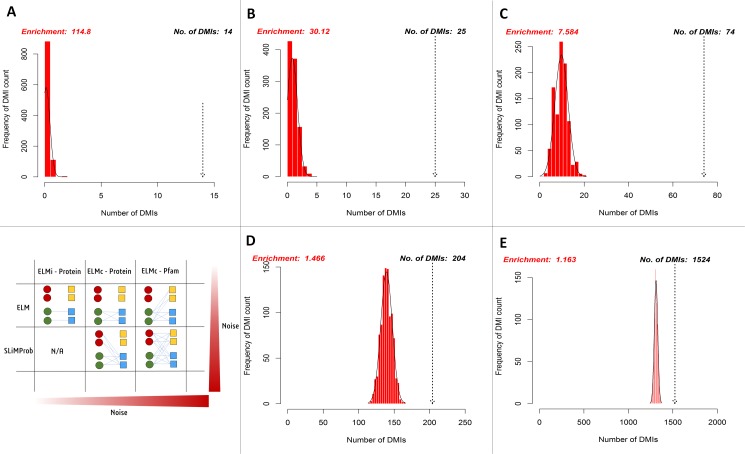
Many important cellular processes involve protein-protein interactions (PPIs) mediated by a Short Linear Motif (SLiM) in one protein interacting with a globular domain in another. Despite their significance, these domain-motif interactions (DMIs) are typically low affinity, which makes them challenging to identify by classical experimental approaches, such as affinity pulldown mass spectrometry (AP-MS) and yeast two-hybrid (Y2H). DMIs are generally underrepresented in PPI networks as a result. A number of computational methods now exist to predict SLiMs and/or DMIs from experimental interaction data but it is yet to be established how effective different PPI detection methods are for capturing these low affinity SLiM-mediated interactions. Here, we introduce a new computational pipeline (SLiM-Enrich) to assess how well a given source of PPI data captures DMIs and thus, by inference, how useful that data should be for SLiM discovery. SLiM-Enrich interrogates a PPI network for pairs of interacting proteins in which the first protein is known or predicted to interact with the second protein via a DMI. Permutation tests compare the number of known/predicted DMIs to the expected distribution if the two sets of proteins are randomly associated. This provides an estimate of DMI enrichment within the data and the false positive rate for individual DMIs. As a case study, we detect significant DMI enrichment in a high-throughput Y2H human PPI study. SLiM-Enrich analysis supports Y2H data as a source of DMIs and highlights the high false positive rates associated with naïve DMI prediction. SLiM-Enrich is available as an R Shiny app. The code is open source and available via a GNU GPL v3 license at: https://github.com/slimsuite/SLiMEnrich. A web server is available at: http://shiny.slimsuite.unsw.edu.au/SLiMEnrich/.
许多重要的细胞过程涉及蛋白质-蛋白质相互作用(PPI),这种相互作用由一种蛋白质中的短线性基序(SLiM)介导,该基序与另一种蛋白质中的球状结构域相互作用。尽管这些结构域-基序相互作用(DMI)很重要,但它们通常亲和力较低,这使得通过经典实验方法(如亲和下拉质谱法(AP-MS)和酵母双杂交法(Y2H))来识别它们具有挑战性。因此,DMI在PPI网络中通常代表性不足。现在有许多计算方法可以从实验相互作用数据中预测SLiM和/或DMI,但不同的PPI检测方法在捕获这些低亲和力的SLiM介导的相互作用方面的有效性尚未确定。在这里,我们引入了一种新的计算流程(SLiM-Enrich),以评估给定的PPI数据源捕获DMI的能力,从而通过推断,评估该数据对于SLiM发现的有用性。SLiM-Enrich在PPI网络中查询相互作用的蛋白质对,其中第一种蛋白质已知或预测通过DMI与第二种蛋白质相互作用。置换检验将已知/预测的DMI数量与如果两组蛋白质随机关联时的预期分布进行比较。这提供了数据中DMI富集的估计以及单个DMI的假阳性率。作为一个案例研究,我们在一项高通量Y2H人类PPI研究中检测到显著的DMI富集。SLiM-Enrich分析支持将Y2H数据作为DMI的来源,并突出了与单纯DMI预测相关的高假阳性率。SLiM-Enrich以R Shiny应用程序的形式提供。代码是开源的,可通过GNU GPL v3许可证在以下网址获得:https://github.com/slimsuite/SLiMEnrich。还有一个网络服务器,网址为:http://shiny.slimsuite.unsw.edu.au/SLiMEnrich/。